Research Article
In silico Characterization of Rhizome coding genes in Ginger and Bamboo
Dhanalakshmi T1, Geethalakshmi S2* and Barathkumar S3
1Department of Microbiology, Nehru Arts and Science College, India
2Department of Biotechnology, Nehru Arts and Science College, India
3Department of Biotechnology, Karpagam Academy of Higher Education, India
Corresponding author: Geethalakshmi S, Department of Biotechnology, Nehru Arts and Science College, Coimbatore, Tamilnadu, India, Tel no: +91-9952411764, E-mail: s.geethalakshmi@gmail.com
Citation: Dhanalakshmi T, Geethalakshmi S, Barathkumar S. In silico Characterization of Rhizome coding genes in Ginger and Bamboo. J Plant Sci Res. 2018;5(1): 177.
Copyright © Dhanalakshmi T, et al. 2018. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Plant Science & Research | ISSN: 2349-2805 | Volume: 5, Issue: 1
Submission: 26/02/2018; Accepted: 28/03/2018; Published: 30/03/2018
Abstract
Rhizome is an underground, horizontal plant stem which helps in propagation of the plants. Growth of rhizome may be facilitated by the genes that are involved in axillary bud initiation. The present study aims at characterizing the genes that are involved in development of rhizome in ginger (Zingiber officinale) and bamboo (Bambusoideae) using in silico tools. The initial dataset was collected from the plant databases and BLASTed against the standard data set. The resulting sequences were subjected to physical parameters, secondary and tertiary structure prediction. The results will form a foundation for analyzing the genetic characters of rhizome which can be extended further for transgenic plant development which has a rich vegetative propagation.
Introduction
The primary storage organ of many perennial grass species is the rhizome which is an underground, horizontal plant stem. It also serves as a primary character for persistence of the plant species [1]. The advantage of rhizome in a plant is it helps in propagation of the plant; whereas the disadvantage is the rhizome in the weeds does not permit the plant to be eradicated completely [2]. Rhizomes were originally identified in plants like ginger (Zingiber officinale) and turmeric (Curcuma longa) based on their ability to develop into a new plant from the buds. The study then extended to grass family where bamboo (Bambusoideae), Sorghum bicolor and Oryza were taken as model plants for the EST analysis [3]. Though several studies has been carried out to characterize the genes involved in propagation of rhizome, due to the polyploidy nature of these perennial plants, identification of full length EST using in vitro techniques is still under progress.
Plant rhizomes originate from axillary buds on the most basal portion of the seedling shoot and hence genes involved in plant axillary bud initiation and outgrowth may contribute to rhizome development and growth [4]. Identification of those genes and its expression can pave way for isolating and using them for transgenic plant development so that plants having a rich vegetative propagation can be developed. This property can be used to protect RET (Rare, Endangered, Threatened) plant varieties.
The present study aims at characterizing the rhizome coding genes from ginger (Zingiber officinale) and bamboo (Bambusoideae) in silico which will provide a base for gaining knowledge about the role and expression of the genes involved in plant propagation via rhizome. The plants considered for the study are not only considered as traditional medicine but also have great economic importance [5].
Materials and Methods
Data set collection and characterization
Rhizome coding nucleotide sequences of Ginger and Bamboo were downloaded from National Centre for Biotechnology (NCBI; www.ncbi.nlm.nih.gov/) for computational analysis [6]. Using the BioEdit tool version 7.0.9 (http://www.mbio.nscu.edu/Bioedit) [7], the downloaded sequences were subjected to six frame translation and the protein sequences were analyzed using PROSITE program in ExPASy server (http://prosite.expasy.org/scanprosite) to locate the characteristic domains specific to the sequences [8,9]. The nucleotide and its deduced protein sequence of the test rhizome coding genes were subjected to nucleotide and amino acid composition analysis using BioEdit version 7.1.3.0 (http://www.mbio.nscu.edu/Bioedit).
Physical parameters prediction
To get more information about the physical nature of the identified rhizome coding genes, protein sequences of predicted genes were subjected to various physical properties analysis such as pI, molecular weight and GRAVY (Grand Average Hydropathy) using PROTPARAM online software was used through the website http://expasy.org/ tools/protparam.html [10].
Secondary structure prediction
The secondary structure of the predicted protein sequences were determined by SOPMA (self optimized prediction method) which is a versatile tool for secondary structure prediction (http://npsa-prabi.ibcp.fr/) [11].
Three dimensional structure prediction using i-TASSER
For the three dimensional structure prediction of the test rhizome coding genes from bamboo and ginger, the iterative threading assembly tool i-TASSER was used (http://zhanglab.ccmb.med.umich.edu/I-TASSER) [12]. The stereochemical properties of the predicted three dimensional structures were confirmed by Ramachandran plot.
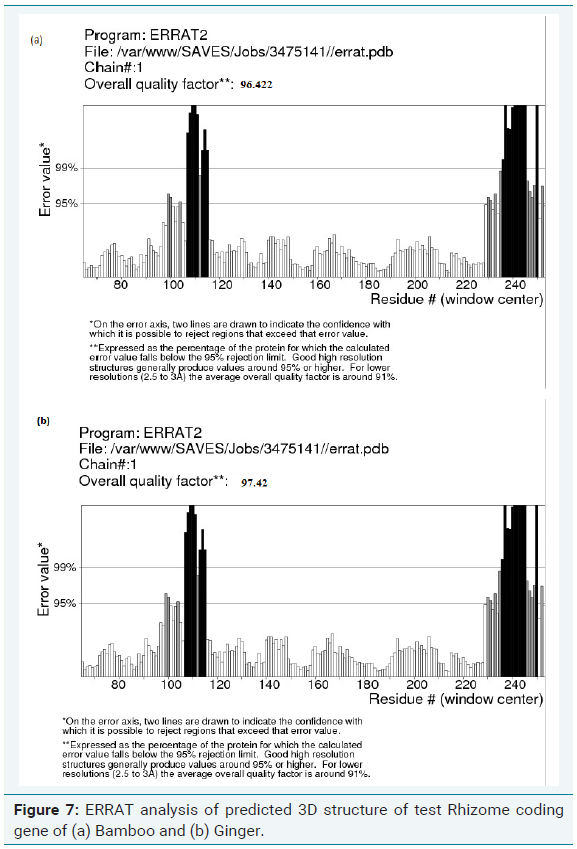
Errat analysis of modelled structure
ERRAT is a program for verifying protein structures determined by crystallography. Error values are plotted as a function of the position of a sliding 9-residue window. The error function is based on the statistics of non-bonded atom-atom interactions in the reported structure (compared to a database of reliable high-resolution structures). According to the analysis by ERRAT, the final model is significantly improved relative to the initial model. The method is sensitive to smaller errors than 3-D Profile analysis [13].
Results and Discussion
Nucleic acid analysis
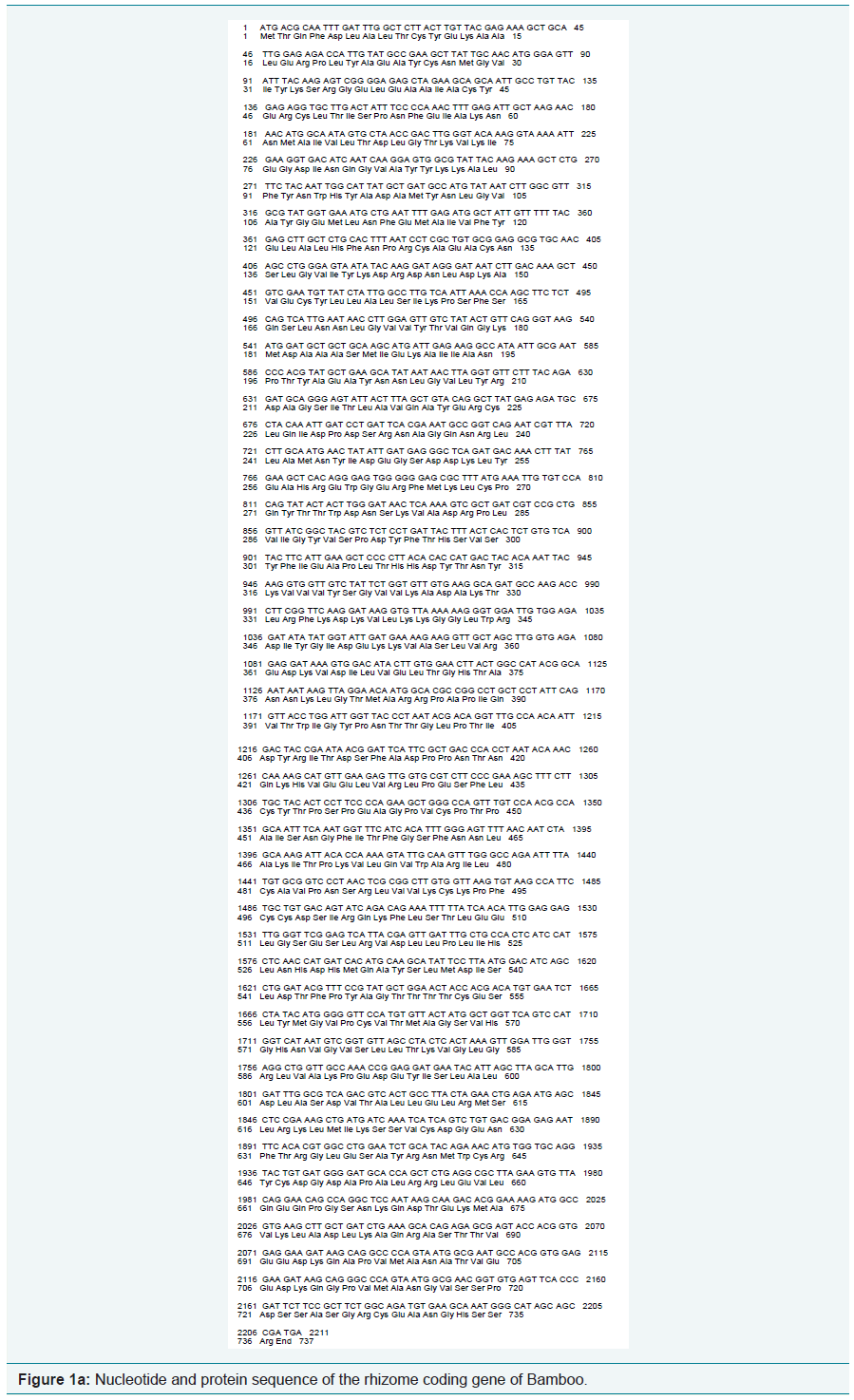
Using the software BIOEDIT, predicted gene sequences of the rhizome of Ginger and Bamboo were subjected to nucleotide analysis (Figure 1a and 1b). According to this, the lengths of the isolated gene were 2211 bp and 339 bp respectively. The number of amino acids coded by the genes was 736 and 112 respectively. The nucleotide number and molecular percentage of the gene sequences are shown in Table 1.

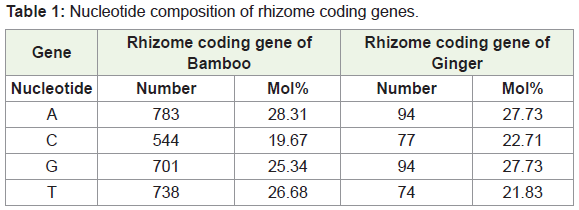
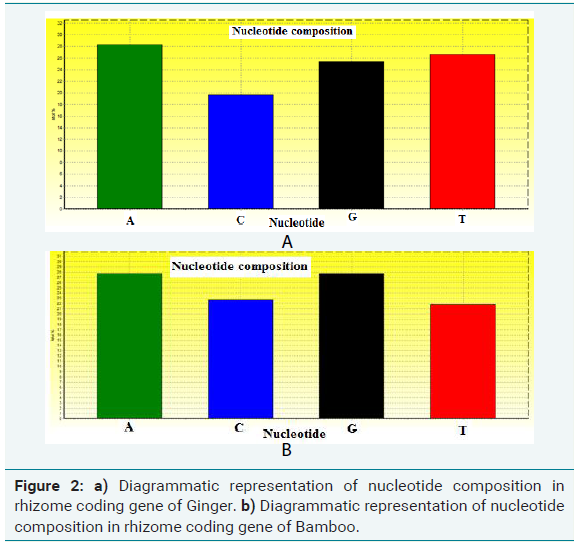
Nucleotide composition of rhizome coding gene in bamboo (Figure 2a)
DNA molecule: Gene 1
Length = 2211 base pairs
Molecular Weight = 668964.00 Daltons, single stranded
Molecular Weight = 1342173.00 Daltons, double stranded
G+C content = 44.87%
A+T content = 55.13%
Nucleotide composition of rhizome coding gene in ginger (Figure 2b)
DNA molecule: Gene 2
Length = 339 base pairs
Molecular Weight = 102980.00 Daltons, single stranded
Molecular Weight = 206140.00 Daltons, double stranded
G+C content = 50.44%
A+T content = 49.56%
Protein analysis-primary structure
The physical parameters of the identified protein sequences were subjected to PROTPARAM analysis at ExPASy server. Based on the parameters predicted, the identified protein sequences were stable and the GRAVY score indicated that the proteins were hydrophilic in nature. This result indicated that the protein has a good interaction with water in the soil which is essential for the enlargement of rhizome and nitrogen translocation [14]. Accumulation of nitrogen in the nodules and rhizome region of plants is mediated by movement of water using the principle of hydraulic lift, which means in the absence of water, the movement of nitrogen is unidirectional and only in the presence of sufficient water the nitrogen gets accumulated throughout the rhizome.
Amino acid analysis
Amino acids were analyzed by using BIOEDIT software, and the composition of the identified protein sequences was depicted in Table 2a and 2b. The amino acid composition of the protein sequences is indicated diagrammatically in Figure 3a and 3b.
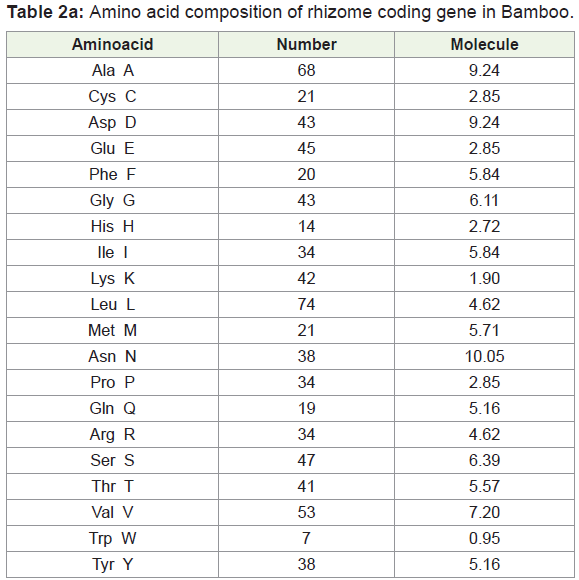
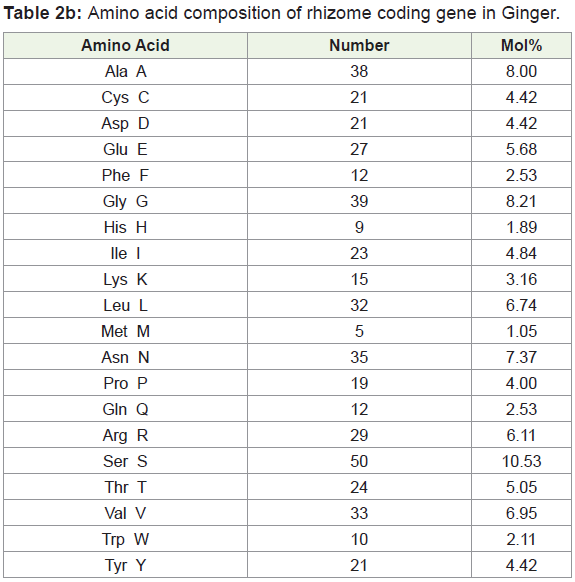
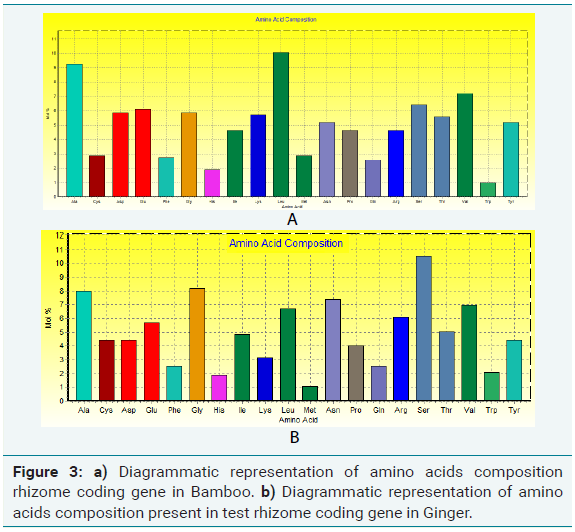
Amino acid composition analysis of Bamboo rhizome protein showed 49.4% of hydrophobic (Gly, 5.84%, Ala, 9.24%; Val, 7.20%; Leu 10.05%; Ile, 4.62%; Pro, 4.62%; Phe, 2.72%; Trp, 0.95%; Met, 2.85%), amino acid residues. Based on the secondary structure predictions, 36.58% of α- helices, 0.00% 310 helix, 18.75% B-turn, and 16.51% contributed the overall helical conformation of Bamboo rhizome coding gene.
Amino acid composition analysis of Ginger rhizome protein showed 49.4% of hydrophobic (Gly, 8.21%, Ala, 8.00%; Val, 6.95%; Leu 6.74%; Ile 4.84%; Pro, 4.00%; Phe, 2.53%; Trp, 2.11%; Met, 1.05%) amino acid residues. Based on the secondary structure predictions, 6.25% of α- helices, 0.00% 310 helix, 18.75% β-turn contributed the overall helical conformation of ginger rhizome coding gene.
From the amino acid composition, it was clear that the identified proteins are capable of forming α helices interconnected by loops. So, the protein can be classified under the helical protein category.
Prosite analysis
Prosite analysis of test protein sequence of rhizome coding gene in bamboo and ginger revealed the presence of two domains:
(a) Six TPR domains which is Tetratricopeptide Repeat (TPRs) (Figure 4a), a degenerate 34-amino acid repeated motif that is widespread among all organisms. In the cell, TPR containing proteins are localized in a variety of subcellular compartment, including the nucleus, the cytoplasm and mitochondria. Processes involving TPR proteins include cell-cycle control, transcription repression, stress response, protein kinase inhibition, mitochondrial and peroxisomal protein transport and neurogenesis. TPR repeats mediate proteinprotein interactions and the assembly of multiprotein complexes. The smallest functional unit that is widely used appears to be three tandem-TPR motifs
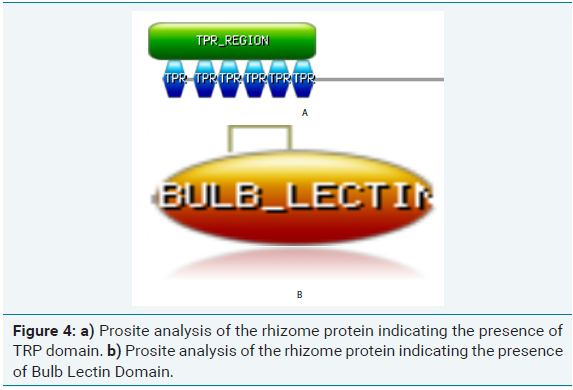
(b) Bulb Lectin super-family domain (Figure 4b) which occurs commonly in Amaryllidaceae, Orchidaceae and Aliaceae. The domain contained a ~115-residue-long domain whose overall three dimensional fold is very similar to that of
Although this domain is a mannose-binding lectin in the bulb super-family, curculin is considered as a non-functional mannosebinding protein devoid of mannose-binding activity.
Secondary structure prediction
For the secondary structure analysis of bamboo rhizome, SOPMA tool was used. The results revealed that the identified protein was predominantly α-helical protein, which mainly consisted of α -helices (41.17%) and random coils (29.62%), extended stand (20.65%) (Figure 5a).
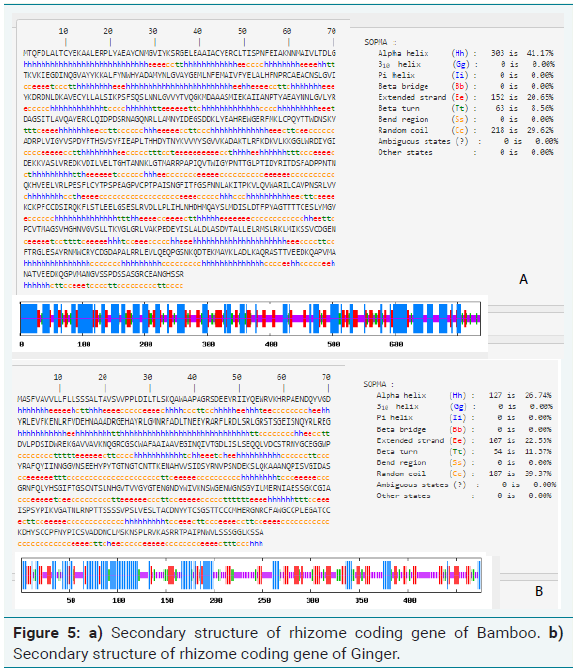
Similarly, SOPMA analysis of ginger rhizome gene revealed that was predominantly α-helical protein, which mainly consisted of α -helices (26.74%) and random coils (39.62%), extended stand (22.53%) (Figure 5b). The secondary structure prediction is in line with the amino acid composition analyzed.
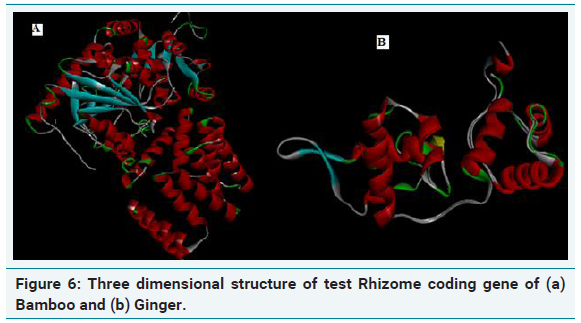
Three dimensional structure of rhizome coding genes
The quality of the model generated was checked by ProSA, PROCHECK and Verify-3D and Ramachandran plot was constructed (Figure 6a and 6b). The R-plot indicated that 75% of the input residues were present in the most favourable region, 17.8% in additional allowed region and 6.7% in generously allowed region. Only one residue (0.5%) was present in disallowed region indicating the overall stereo chemical stability of the predicted structure. PROCHECK, Verify-3D and ERRAT results also confirmed the stability and reliability of the predicted 3D structure (Figure 7a and 7b). From this result, it is clear that the protein is highly stable with stabilized three dimensional structure which is highly hydrophilic in nature encompassing helical domain, which is the major domain which contributes to the function of the protein.
Summary
Rhizome families are well-known for its medicinal and economic significances. Many species are used as sources of indigenous medicines, vegetables, food flavours, spices, dyes, condiments as well as ornamentals. This family is widely known for its broad range of pharmacological activities [15]. To study on its compounds offers many opportunities to investigate the various functions and prospects in various pharmaceutical studies. The present study forms the basis for understanding the structure of rhizome coding gene’s structure from its sequence level; it will become more evident about its potential from the bioactivities, in reviewing sequence to structure to functions in wide group of rhizome families. Detailed study of the functional role of rhizome coding protein may be utilized for regeneration and protection of RET plants.
References
- Hu F, Wang D, Zhao X, Zhang T, Sun H, et al. (2011) Identification of rhizome-specific genes by genome-wide differential expression analysis in Oryza longistaminata. BMC Plant Biol 11: 18.
- Jang CS, Kamps TL, Skinner DN, Schulze SR, Vencill WK, et al. (2006) Functional classification, genomic organization, putatively cis-acting regulatory elements, and relationship to quantitative trait loci, of sorghum genes with rhizome-enriched expression. Plant Physiol 142: 1148-1159.
- Koo HJ, McDowell ET, Ma X, Greer KA, Kapteyn J, et al. (2013) Ginger and turmeric expressed sequence tags identify signature genes for rhizome identity and development and the biosynthesis of curcuminoids, gingerols and terpenoids. BMC Plant Biol 13: 27.
- Gizmawy I, Kigel J, Koller D, Ofir M (1985) Initiation, orientation and early development of primary rhizomes in Sorghum halepense (L.) Pers. Annals of Botany 55: 343-350.
- Wang K, Peng H, Lin E, Jin Q, Hua X, et al. (2010) Identification of genes related to the development of bamboo rhizome bud. J Exp Bot 61: 551-561.
- NCBI Resource Coordinators (2016) Database resources of the national center for biotechnology information. Nucleic Acids Res 44: D7-D19.
- Hall T, Biosciences I, Carlsbad C (2011) BioEdit: An important software for molecular biology. GERF Bull Biosci 2: 60-61.
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, et al. (2003) ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31: 3784-3788.
- de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, et al. (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res 34: W362-W365.
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, et al. (2005) Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook pp: 571-607.
- Geourjon C, Deléage G (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 11: 681-684.
- Yang J, Yan R, Roy A, Xu D, Poisson J, et al. (2015) The I-TASSER Suite: protein structure and function prediction. Nat Methods 12: 7-8.
- Colovos C, Yeates TO (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 2: 1511-1519.
- de Kroon H, van der Zalm E, van Rheenen JW, van Dijk A, Kreulen R (1998) The interaction between water and nitrogen translocation in a rhizomatous sedge (Carex flacca). Oecologia 116: 38-49.
- Duke J (2012) Handbook of legumes of world economic importance. Springer Science & Business Media pp: 346.