Review Article
Comprehending Human Disorders: Focus on Reactive Oxygen Species
RenuYadav, Kritika Raj and Surajit Sarkar*
Corresponding author: Dr. Surajit Sarkar, Assistant Professor, Department of Genetics, University of Delhi, India, Fax: +91-11-24112761,; E-mail: sarkar@south.du.ac.in
Citation: Yadav R, Raj K, Sarkar S. Comprehending Human Disorders: Focus on Reactive Oxygen Species. J Cell Sci Molecul Biol. 2014;1(3): 110.
Copyright © 2014 Surajit Sarkar et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Submission: 04/09/2014; Accepted: 08/12/2014; Published: 09/12/2014
Abstract
Reactive Oxygen Species (ROS) are highly reactive oxygen moieties which result in the cellular state of oxidative stress, either due to their overproduction or by imbalance between cellular oxidants and antioxidant system. Mitochondria is the main source and site of ROS production inside a cell and accountfor both - electron transport chain (ETC) and non-ETC dependent sources. Interestingly, a threshold level of ROS is essential for cellular functioning, nevertheless, any unexpected increase in its level poses detrimental effects on the cells by oxidizing biomolecules such as proteins, lipids, nucleic acids etc. Although cellular machinery is well equipped with both enzymatic and non-enzymatic antioxidant defense systems, cellular injury and aging due to generation of ROS is inevitable in all living systems. Higher levels of ROS are thus been commonly associated with onset of several human diseases such as neurodegenerative disorders, cancer, infertility etc. Present review attempts to provide a brief account of ROS and their regulation in cellular systems along with their potential involvement in predisposition of some fatal human diseases.
Keywords: ROS; Mitochondria; Oxidative stress; Neurodegeneration; Cancer; Infertility
Introduction
Establishment of aerobic conditions by photosynthesizing archae and eubacteria marked one of the most significant changes that the earth’s atmosphere underwent millions of years ago. This grand event facilitated evolution of organisms which could utilize oxygen for energy metabolism and survival in a process called aerobic respiration. Interestingly, in spite of being much more efficient than anaerobic respiration, aerobic respiration offers several opportunities for oxygen leakage and results in the consequent production of Reactive Oxygen Species (ROS). The term ROS, in effect, refers to all those free radicals which are composed of oxygen molecules. Overwhelming levels of ROS either due to excessive ROS production or inadequate action of antioxidants leads to a cellular condition termed as “oxidative stress”. This review focuses on the involvement of ROS and associated oxidative stress in predisposition and progression of some of the major human disorders.
Sources and physiological implications of ROS
Mitochondria represent the major site of ROS production in a cell. One of the first reports to suggest generation of ROS in the mitochondria was put forward by Jensen in 1966 [1]. It was observed that a portion of the total oxygen taken up by the mitochondria was consumed in a catalase-sensitive reaction and thus, proposed that mitochondrial NADH/succinate oxidation might be coupled with hydrogen peroxide (H2O2) production [1]. Later in 1972-1973, Britton Chance and co-workers carried out a more generalized study and instigated the field of mitochondrial ROS research [2,3]. It is increasingly clear now that under normal physiological conditions, approximately 2% of the electrons leak from the electron transport chain (ETC) and account for ROS production [4]. The major types of ROS found in living animals include superoxide anion (O2•−), hydrogen peroxide (H2O2) and hydroxyl radical (•OH). When an electron encounters an oxygen atom, a reduction reaction takes place and O2•− is formed. O2 •− is considered to be the most importantoxygen free radical and the source of other ROS molecules. O2 •− is readily converted into H2O2 by superoxide dismutase (SOD) enzyme which is in turn converted into •OH in the presence of ferrous (Fe2+) or cuprous (Cu2+) ions [5]. While •OH is highly reactive, H2O2 is morestable and membrane permeant.
There are around eight known sites in the mitochondria which possess the ability to produce O2•−, however, the major sites of O2•− production along the ETC are NADH-ubiquinone oxidoreductase (Complex I) and ubiquinone-cytochrome c oxidoreductase (ComplexIII) [6,7]. Non-ETC sources of mitochondrial ROS production include monoamine oxidase, located in the outer mitochondrial membrane and produces H2O2 as a byproduct of oxidative deamination; glycerol-3-phosphate dehydrogenase (GPDH) located in the mitochondrial matrix and produces O2 •−and α-ketoglutaratedehydrogenase (α-KGDH) which is also located in the mitochondrial matrix and produces both O2 •− and H2O2 under elevated NADPH/ NADP+ ratio i.e. during times of maximum respiration and during availability of calcium.
Out of all these mitochondrial ROS producing hubs, ROS produced into the intermembrane space (by Complex III and GPDH) is of greater relevance than the others since these have comparatively easier access to the cytosol as ROS produced there need to cross only outer mitochondrial membrane whereas others must cross both the inner as well as the outer mitochondrial membrane to access the cytosol. Besides mitochondria, ROS are produced from cytosolic sources as well. These include oxidation reactions of dopamine and xanthine oxidase (XO)-mediated conversion of hypoxanthine to xanthine, and xanthine to uric acid which subsequently generates O2•−. Furthermore, elevated levels of calcium in neuronal cells are also associated with ROS production which initiates signaling events in the cytosol. Occurrence of certain cell types such astrocytes, phagocytes (microglia), lymphocytes etc. also contribute to the rate of ROS production. For example, NADPH oxidase (Nox) located in the cell membrane of CNS-resident astrocytes and phagocytes (microglia) and 5-lipoxygenase present in lymphocytes serve as inducible sources of ROS, especially O2•− [8].
Mitochondrial ROS production is dependent on a number of factors; the redox state of ETC being the most important one. The tendency to generate O2 •− free radicals increases when the ETC electron carriers become more reduced due to inhibition of electron transfer. Another major determinant of ROS production in the mitochondria is the proton motive force (pmf) which is generated as protons are expelled into the intermembrane space from the matrix while they are transferred from complex I to complex IV. Greater the pmf, more is the ROS production. ROS molecules have been implicated in various signaling pathways. In this context it is important to note that the sub-cellular localization of the mitochondria affects the strength of ROS signaling in the cell. Closer the site of ROS production and operation, greater is the efficiency of signaling [9]. ROS signaling is involved in the regulation of a variety of physiological processes like cell migration and proliferation, autophagy, immune responses and adaptation to hypoxia or oxidative stress conditions. Most of thesefunctions are mediated via derivatives of O2 •− especially H2O2 and •OH. For instance, activated macrophages and neutrophils produce high levels of O2 •− which coalesce with other ROS molecules in order to destroy the invading pathogens. This phenomenon is known as the “oxidative burst” [10]. O2 •− is also suggested to play an age-dependent role in the regulation of synaptic plasticity, memory formation and learning [11]. H2O2 acts by oxidizing thiol groups (-SH-) on cysteine residues thereby inactivating the target proteins in most of the cases. For example, H2O2 inactivates the Protein Phosphatase 2A (PP2A) at the cysteine residue to activate the Akt pathway for promotion of cell survival [12-14]. •OH has been demonstrated to regulate cell physiology by stimulating guanylatecyclase to produce cGMP which acts as a “secondary messenger” in the cell and activates the transcription factor Nuclear Factor Kappa-light-chain-enhancer of activated B cells (NF-κB) with the help of H2O2 [5]. NF-κB signals play an essential role in regulating cellular immune responses against any kind of infection.
Intriguingly, ROS exhibit both beneficial as well as deleterious effects. Since ROS are highly reactive in nature, excessive production or accumulation of ROS (oxidative stress, as discussed above) most often proves to be detrimental for the cell. ROS can oxidise a variety of biomolecules including lipids, proteins, DNA as well as RNA. Lipid peroxidation causes membrane leakage [15], amino acid oxidation, protein misfolding and dysfunction [16]. ROSinduced DNA damage leads to the production of oxidised bases (e.g., 8-hydroxy-deoxyGuanosine; 8-OHdG), oxidised sugar fragments (e.g., deoxyribonolactone), oxidised abasic sites (e.g., apurinic sites) and single-strand and double-strand breaks, all of which interfere with gene transcription [17]. RNA being single stranded in nature is more susceptible to the adverse effects of ROS as compared to DNA. Mutilation of protein-coding or non-coding RNA leads to interruption of protein synthesis and dysregulation of gene expression respectively [18]. An accumulative and simultaneous exposure to ROS may lead to major cellular damage and cell death. In view of above, several antioxidant systems, both enzymatic and non-enzymatic, have been employed to maintain the physiological levels of cellular ROS. Some of the major enzymatic antioxidants include superoxide dismutase (SOD), glutathione peroxidase (GPx), thioredoxin reductase (TR) and catalase [19]. SOD scavenges O2•− by converting it into O2 and H2O2. Subsequently, activities of generated H2O2 facilitated by GPx and TR, which finally lead to production of H2O and O2. Non-enzymatic antioxidants include exogenous sources of defense like ascorbic acid (vitamin C), α-tocopherol (vitamin E), glutathione (GSH) and flavonoids [20].
Despite all these lines of protective measures, aging is a biological phenomenon which is inevitably associated with production and accumulation of cellular ROS. The observation dates back to the 1950s when Harman first proposed the free radical theory of aging which suggested that accumulation of ROS-induced cellular damage is the major determinant of longevity [21]. The theory was later modified to the mitochondrial free radical theory of aging as mutations in the mitochondrial DNA (mtDNA) were perceived as the foundation of aging phenomenon [22,23]. According to this theory, mutation(s) in mtDNA result in impaired functioning of respiratory chain and subsequent production of ROS. This phenomenon facilitates additional damage in mtDNA by forming a vicious circle whichamplifies the damage, and consequently, causes the cells to age [22,23]. Several studies have demonstrated that aging is marked by an increase in mtDNA mutations, impaired ETC functions, poor mitochondrial membrane integrity and dysregulated levels of cytosolic calcium [24]. One of the most direct experimental evidences that suggest an active role of mitochondrial ROS in longevity was provided by studies carried out on mice wherein overexpression of catalase targeted to the mitochondria resulted in a significant extension in lifespan [25]. Interestingly, similar overexpression of catalase targeted to the nucleus or to the peroxisome, its native location within the cell, did not have any noteworthy effect on murine lifespan [25].
Role of ROS in human diseases
Neurodegenerative disorders
Several age-related disorders are associated with elevated levels of oxidative stress. The human brain is one organ that is particularly sensitive to the hazardous effects of ROS. This is primarily due to the fact that the brain has a high respiration rate but a weak antioxidant framework. In addition, brain cells are rich in dopamine and polyunsaturated fatty acids which are highly prone to oxidation by ROS. High levels of iron in the brain acts as pro-oxidant and promotes oxidative stress-induced damage in neurons. Further, neurons lack replication-coupled repair mechanisms which lead to persistent oxidative genome damage and cell death. Moreover, since the neurons are differentiated and post-mitotic cells; loss of “sick” neurons are not compensated for, and thus, the damage becomes much more prominent [17,26]. This leads to the predisposition/ development of a number of neurodegenerative diseases like Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington disease (HD), Amyotrophic lateral sclerosis (ALS) etc. (Figure 1). A common feature shared by all of these diseases is their late-onset nature, which may be attributed to the gradual accumulation of ROS mediated damages with age and a sudden manifestation of the disease phenotypes represent the moment when the destruction crosses the threshold mark.
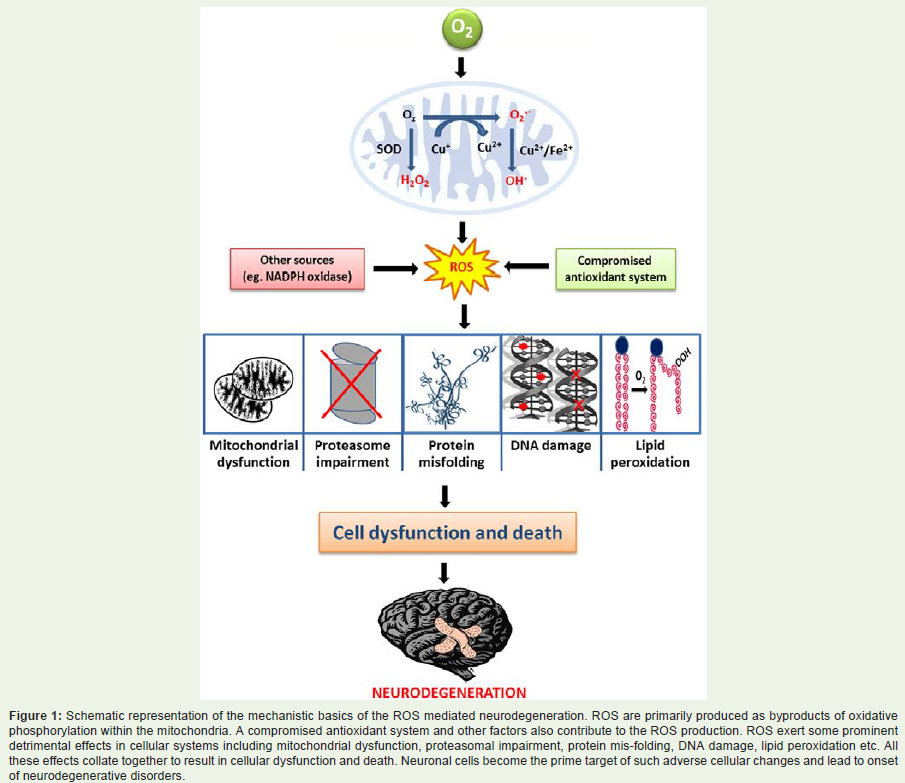
Figure 1: Schematic representation of the mechanistic basics of the ROS mediated neurodegeneration. ROS are primarily produced as byproducts of oxidative phosphorylation within the mitochondria. A compromised antioxidant system and other factors also contribute to the ROS production. ROS exert some prominent detrimental effects in cellular systems including mitochondrial dysfunction, proteasomal impairment, protein mis-folding, DNA damage, lipid peroxidation etc. All these effects collate together to result in cellular dysfunction and death. Neuronal cells become the prime target of such adverse cellular changes and lead to onset of neurodegenerative disorders.
AD is the most common form of dementia worldwide. It isassociated with the formation of neurofibrillary tangles (NFT) and neural plaques (NP) in the brain [27]. While NFTs are rich in hyper phosphorylated Tau protein, NPs are abundant in amyloid β protein. ROS have been shown to be actively involved in Tau phosphorylation [28]. Once phosphorylated, Tau becomes more prone to modification by the products of oxidative stress (e.g., 4-hydroxy-2-nonenal; HNE) resulting in conformational changes in the protein and promotion of tangle formation [29,30]. Similarly, studies in neuronal cell lines have demonstrated that elevated levels of H2O2 result in an increased level of intracellular Aβ [31]. Higher levels of Aβ activate Nox which subsequently leads to production of O2 • and conversion of molecular oxygen into H2O2 by reducing cellular Fe2+ and Cu2+ [31]. Aβ accumulation also causes an uprise in the calcium levels of the cell that finally results in membrane disruption by formation of calcium-conducting pores [31]. All these events further aggravate the oxidative stress on the neuronal cells [32]. Nuclear and mitochondrial DNA isolated from neurons of the affected areas of the brain of AD patients were found to be abundant in oxidised bases, especially 8-OHdG which is considered to be a major marker of oxidative damage to the DNA [33]. Interestingly, a recent study has further demonstrated that enhanced expression of mitochondrial catalase (mCAT) in mice model of AD resulted in a significant decrease in Aβ toxicity and cellular oxidative damages [34]. This suggests a direct role of mitochondrial oxidative stress in AD pathology. Moreover, it appears that mitochondria targeted antioxidant strategies could be advanced further to design novel therapeutic measures for AD.
PD is another chronic neurodegenerative disorder which is caused by the loss of dopamine-generating neurons in the basal ganglia of the brain [35]. Neuronal degeneration in this case is stimulated by the deposition of cytoplasmic inclusions called Lewy bodies whose major constituent includes the ubiquitin-bound α-synuclein protein (α-Syn). Posttranslational modifications of α-Syn, mainly oxidation by HNE, have been indicated to promote its oligomerisation [36]. Oxidative stress also results in microglial senescence and compromised protein clearance by the ubiquitin proteasome system [37]. This phenomenon has been proposed to be a consequence of intracellular accumulation of iron [37]. In addition, it also causes enhancement in the level of 8-OHdG, indicating stressinduced DNA damage [33]. Moreover, several PD-causing mutations have been found to be associated with PTEN-induced putative kinase 1 (PINK1), DJ-1, leucine-rich repeat kinase 2 (LRRK2) and α-Syn itself, all of which are either mitochondrial proteins or associated with it in a way that links mitochondria to oxidative stress [38,39]. Further, dopamine oxidation by monoamine-oxidase-B induces excessive production of all three major types of ROS [40]. Reduced level of GSH is another characteristic feature of PD, with the extent of depletion being directly proportional to the severity of the disease [41]. As noted in case of AD, mCAT over expression in mouse brains have protective effect against a neurotoxin, 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine (MPTP) induced PD as well [42].
HD is a triplet repeat-expansion disorder that is caused by an autosomal dominant mutation in the gene coding for the hunting tin (Htt) protein. Mutant Htt (mHtt) is yet another aggregationprone protein which interacts with several other cellular proteins to result in the formation of intranuclear aggregates called inclusion bodies (IB) [43]. The size and conformation of constituent proteins of the aggregates have been shown to be modified upon exposure to oxidative stress [44]. In addition, mHtt is believed to inhibit mitochondrial oxidative phosphorylation resulting in increased ROS production and an augmented oxidative challenge [45]. Unlike other cases, oxidative DNA damage has not been extensively observed in HD patients, however, significant increase in the levels of lipid peroxidation has been considered as a major phenomenon that advocates the role of oxidative stress in HD pathology [34].
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder which causes damage to the motor neurons in the brain. Affected motor neurons have been demonstrated to accumulate proteinaceous inclusions both in the cell body and in axons. These inclusion bodies sequester SOD1 enzyme quite often which leads to compromised antioxidant system of the cell and makes it susceptible to oxidative assault [38]. Approximately 20% of the ALS cases also exhibit mutations in SOD1 gene. Subsequently, elevated levels of lipid peroxidation and that of 8-OHdG can be commonly observed in ALS patient samples [34]. Disrupted mitochondrial membrane potential and high cytosolic calcium levels are other characteristic features of ALS [8].
Human cancers
Cancer is one of the most deadly diseases leading to the highest number of demises worldwide. Onset of cancer is a result of uninhibited cell growth and proliferation triggered due to mutations in specific genes/DNA by carcinogens. Carcinogens could be physical (ultraviolet radiation), chemical (tobacco smoke, arsenic) or biological (viruses, bacteria) in nature. Unremitting exposure to above factors may cause gain of function mutations in proto-oncogenes transforming them to oncogenes and loss of function mutations in tumor suppressor genes, thereby perturbing the expression pattern of the genes [46]. Altered expression of these genes leads to increased cell proliferation and transformation of cells into malignant tumorous cells. Such cells loose contact inhibition and become resistant to apoptosis and also stop responding to the signals which arrest cellular growth and proliferation [47]. Angiogenesis (formation of new blood vessels from the existing ones) induced due to the secretion of various growth factors by the tumorous cells is a key characteristic of any cancer type [48].
Cancer etiology has been found to be multi factorial which depends upon factors such as age, genetic makeup, environment, life style etc. All these risk factors may instigate the development of cancer by tampering with the ROS levels inside the cell and causing oxidative stress [49]. As discussed above, ROS play an intricate role in regulating various cellular processes including metabolism, signaling pathways, gene expression, cell proliferation and apoptosis. Therefore, an imbalance in ROS levels disturbs the cellular homeostasis and aggravates the cancer pathology. In this context it is important to note that ROS act like a double-edged sword in case of cancer development. On one side, ROS may transform various protooncogenes to oncogenes and activate various transcription factors involved in cellular proliferation; on contrary they could also control expression of several tumor suppressor genes which regulate DNA damage repair, cell cycle arrest and apoptosis [50]. In view of above, ROS function as both - positive as well as negative regulator of cell proliferation and persistence. Cancer cells utilize this dual property exhibited by ROS in encouraging cell growth, survival and inducing genomic instability, which are some of the key features of cancerous cells [51].
ROS in regulation of oncogenes and transcription factors
There are several evidences which suggest a potential link between ROS and oncogenic transformations. Although the operating mechanism behind activation of oncogenes by ROS is not well understood, however, ROS mediated DNA damage has been proposed to be a major factor. It has been found that constitutive expression of isoforms of Rac and Ras genes leads to oncogenic transformation of fibroblasts, which is further associated with production of superoxides. It was demonstrated that such transformation could be suppressed by treating the fibroblasts with antioxidants [52]. In this context it is important to note that ROS have been demonstrated to up-regulatethe expression of certain oncogenes like Fos and Jun [53]. Subsequently, increased levels of ROS have also been found to be associated with oncogenic transformation of ovarian epithelial cells with H-Ras(V12) and tyrosine kinase Bcr/Abl in cases of hematopoietic cells [54]. In addition to Bcr/Abl, other oncogenes such as Flt3-ITD, Ras and c-Kit have not only been found to be involved in leukemia signaling but have also been observed to induce alteration in ROS homeostasis in various cancer models [55].
In addition to the oncogenes, ROS mediated alteration in expression of various transcription factors have also been observed and linked with cancer. It has been reported that transcription factors such as nuclear factor NF-KB, activator protein-1 (AP-1), hypoxiainducible factor-1a (HIF-1a) and signal transducer and activator of transcription 3 (STAT3) are activated by ROS [56]. Activation of these transcription factors result in expression of genes involved in cellular transformation, inflammation, tumor cell survival, proliferation and invasion, angiogenesis and metastasis [56]. It has been found that even in absence of any physiological trigger, activation of NF-KB could be elicited by ROS, which in turn could be inhibited by several chemically divergent antioxidants [57]. Another study which utilized mouse epidermal JB6 cell lines as model system for skin carcinogenesis has demonstrated that functional inactivation of NF-KB and AP1 could restrict the transformation of the JB6 cells to cancerous cells. A similar inhibition was also observed in JB6 cell lines when activities of NF-KB and AP1 were repressed by antioxidants [58]. Above studies clearly suggest a potential role of ROS in transformation of JB6 cells in tumorous cells.
ROS in regulation of tumour suppressor genes
Similar to the role of ROS in modulating activities of oncogenes and transcription factors, they also play an imperative role in the pathways regulated by tumor suppressor genes. Tumor suppressor genes are known to be involved in regulating various aspects of cell division and other activities [50]. Aberration in the functioning of these genes results in uncontrolled cell growth leading to tumor development. ROS have been found to influence the expression of various tumor suppressor genes such as p53, FoxO, retinoblastoma(RB), p21, p16, breast cancer susceptibility genes 1 and 2 (BRCA1 andBRCA2) etc. [59].
Among these tumor suppressor genes p53 is the most commonly mutated gene found in most of the cancer types. In response to various cellular cues, p53 regularly keeps a tight check on cell cycle arrest, senescence and apoptosis [60]. Oxidative stress causes activation of p53 which in turn performs antioxidant functions [61]. It has been found that p53 is involved in antioxidant defense as it induces the expression of many antioxidant genes like GPx1, mitochondrial SOD2 and mammalian sestrin homologs 1 and 2 [59]. In this context it is important to note that down regulation of p53 results in increased levels of ROS [61]. However, there are incidences where p53 induced enhanced transcription of pro-oxidant genes causes elevation in the levels of intracellular ROS and cell death [62]. Under stress conditions, p53 induces ROS production by triggering the transcription of pro-apoptotic genes like p53-induced gene 3 (PIG3) and PUMA. Therefore, p53 target genes which include both antioxidants and prooxidants, have a conflicting effect on the intracellular ROS. This could be debated as low levels of p53 under normal physiological condition, functions as antioxidant and reduces the ROS levels but under conditions of cellular stress, a high level of p53 facilitates pro-oxidant function resulting in increased accumulation of ROS. Therefore, a concentration dependent role of p53 as a pro oxidant and antioxidant has been proposed [59].
Forkhead box O (FoxO), is another tumor suppressor gene which has been implicated in regulation of various cellular functioning and suggested to be regulated by the status of cellular homeostasis and oxidative stress. Expression and functioning of FoxO is regulated largely by two signaling pathways i.e. insulin signaling and cellular stress signaling, latter one primarily driven by oxidative stress [63]. Under normal growth conditions, activation of insulin pathway results in cytoplasmic translocation of FoxO (Figure 2A). In cytoplasm, FoxO is polyubiquitinated by ubiquitin ligase to persuade the proteasomal degradation (Figure 2A). Cytoplasmic sequestration/ degradation of FoxO inhibits FoxO dependent transcription of target genes and promotes cell growth, survival and proliferation [64,65]. However, during oxidative stress condition, phosphorylation of FoxO by mammalian sterile 20-like kinase (MST1) along with JNK dependent phosphorylation of 14-3-3 chaperone proteins (which mediated sequestration of FoxO in cytoplasm) facilitates its nuclear translocation and activation of target genes [66,67,63] (Figure 2B). In addition to the phosphorylation, other post translational modifications such as acetylation and ubiquitylation also regulate FoxO related activities in response to oxidative stress [68]. Moreover, ROS mediated up-regulation of other transcription factors also alter the expression dynamics of FoxO. One such transcription factor is beta-catenin which directly binds to FoxO to increase its transcriptional activities [69]. All such positive regulators of FoxO thereby enhance its transcriptional activities by sequestering it in the nucleus and inducing the expression of target genes which subsequently results in cell cycle arrest, programmed cell death, DNA repair and oxidative detoxification. Taken together, it appears that under influence of the high levels of ROS, FoxO activates adaptive or apoptotic responses, which in turn inhibit tumor progression [68].
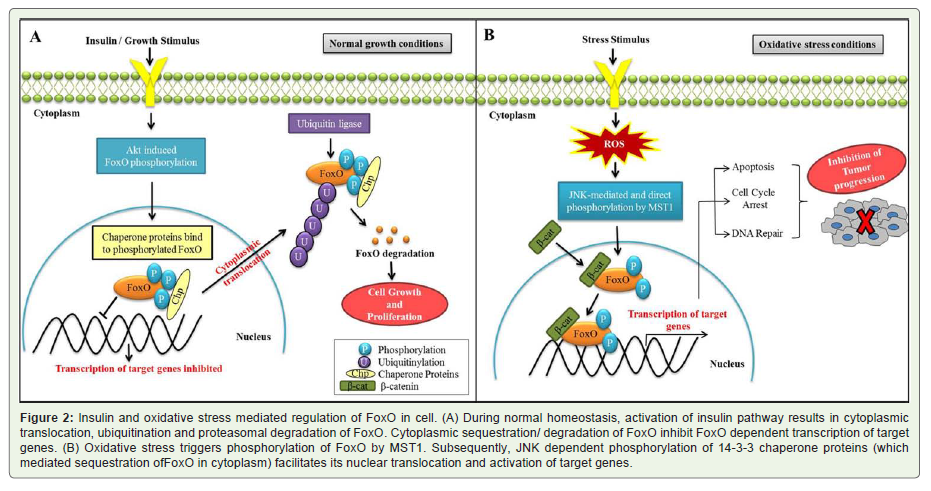
Figure 2: Insulin and oxidative stress mediated regulation of FoxO in cell. (A) During normal homeostasis, activation of insulin pathway results in cytoplasmic translocation, ubiquitination and proteasomal degradation of FoxO. Cytoplasmic sequestration/ degradation of FoxO inhibit FoxO dependent transcription of target genes. (B) Oxidative stress triggers phosphorylation of FoxO by MST1. Subsequently, JNK dependent phosphorylation of 14-3-3 chaperone proteins (which mediated sequestration ofFoxO in cytoplasm) facilitates its nuclear translocation and activation of target genes.
Male infertility
Similar to other fatal diseases like cancer, diabetes, and neurodegenerative disorders; infertility is also seeking lot of concern from the researchers worldwide. There is alarming increase in the number of infertility cases, especially in metropolitan cities.Statistically, around 13-20% of couples around the world are affected by infertility related problems [70]. Of these, male infertility accounts for approximately 30% cases [71].
Oxidative stress has been implicated as one of the major causative factors in male infertility. The first indication of susceptibility of male germ cells towards high levels of ROS was shown by Macleod in 1943. He demonstrated in vitro that human spermatozoa lose their motility in presence of extracellularly generated ROS [72]. Moreover, sperm motility could be rescued by incubating them in the antioxidant rich medium. Later, lipid peroxidative damage to the sperm plasma membrane by ROS was observed to be accountable for the loss of sperm motility [73]. Mammalian spermatozoa are especially susceptible to damage by ROS since their plasma membranes are rich in unsaturated fatty acids which provide necessary fluidity for fertilization and membrane fusion. These unsaturated fatty acids are vulnerable to oxidative stress and once initiated, lipid peroxidation chain reaction decreases membrane fluidity and integrity which subsequently leads to impairments in sperm functioning and male infertility [74,75].
The preliminary cellular sources of ROS in semen are sperm cells. ROS are essential for condensation of chromatin in sperm nucleus, for inducing apoptosis to regulate the number of germ cells and for the proliferation of spermatogonia [76]. Therefore male germ cells produce trivial amounts of ROS from the beginning of spermatogenesis [77]. In mature sperm, ROS aid in capacitation, acrosome reaction, mitochondrial sheath stability and signaling [70]. Another important source of ROS in semen is leukocytes which in general produces around 1000 times more ROS than sperm cells [78]. Leukocytes count generally increase in semen due to infection and environmental factors finally leading to oxidative stress [79].
Since spermatozoa are highly vulnerable to oxidative stress, therefore, semen is laden with protective antioxidant defense system. Antioxidant defense machinery of semen constitutes both enzymatic and non-enzymatic members [79,80]. Antioxidant defense system normalizes oxidative stress encountered by sperm cells during differentiation and maturation in male reproductive tract [81]. Therefore, any fluctuation in antioxidant levels in semen due to changed modern life style or environmental factors induces oxidative stress in sperm cells initiating infertility in men.
Oxidative stress affects the male fertility not only by reducing fertilization capability of sperm but also causes a lot of damage to nuclear and mitochondrial sperm DNA. DNA fragmentation has negative relationship to the sperm quality which includes sperm count, mobility and morphology [81]. Independently, numerous studies have also demonstrated a negative correlation between infertility and DNA damage due to ROS or other factors. Damaged DNA from ejaculates of such infertile men is usually associated with oxidative stress as depicted by the presence of 8-OHdG [82,83]. One of the major consequences of damaged DNA in sperm due to oxidative stress is the increased mutational load on embryo developed due to fertilization of egg and such sperm. This increases the risk of improper embryonic implantation, miscarriage and also facilitates predisposition of diseases such as cancer and other genetic disorders in offspring [84,85]. Therefore, a comprehensive effort should be made to investigate the mechanistic details on ROS and germ cell development in order to design novel therapeutic strategies.
Conclusion and Future Prospects
As discussed earlier, recent times have witnessed the emergence of ROS as one of the major risk factors for development of various human disorders. Intriguingly, ROS in itself are essential for cell survival but their overproduction forces cell to undergo oxidative stress. To evade such conditions, efficient antioxidant systems need to be in place to maintain the normal cellular homeostasis. Any disequilibrium in the cellular pro-oxidants: anti-oxidants system due to biological and/or environmental factors could trigger the process of disease progression. Once disease condition has set in, pathological changes in turn induce production of more ROS, and therefore, from a cause it now becomes a consequence. This vicious circle further worsens the disease conditions and causes development of several additional ailments. In view of above, ROS may serve as an eligible target for therapeutic interventions. Therefore, future research should focus on development of methodologies to prevent production of undesirable ROS. This approach will aid in treatment of oxidative stress related fatal human disorders.
Acknowledgements
Research programmes in laboratory is supported by grants from the Department of Biotechnology (DBT), Government of India, New Delhi; Department of Science and Technology (DST), Government of India, New Delhi, and Delhi University R & D fund to SS. RY is supported by the Senior Research Fellowship (SRF) Professional under DST-INSPIRE program and KR is supported by Junior Research Fellowship (JRF) from the University Grant Commission (UGC), New Delhi.
References
- Jensen PK (1966) Antimycin-insensitive oxidation of succinate and reduced nicotinamide adenine dinucleotide in electron-transport particles. I. pH dependency and hydrogen peroxide formation. Biochim Biophys Acta122: 157-166.
- Boveris A, Oshino N, Chance B (1972) The cellular production of hydrogen peroxide. Biochem J 128: 617-630.
- Boveris A, Chance B (1973) The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J 134: 707-716.
- Chance B, Sies H, Boveris A (1979) Hydroperoxide metabolism in mammalian organs. Physiol Rev 59: 527-605.
- Dröge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82: 47-95.
- Brand MD (2010) The sites and topology of mitochondrial superoxide production. Exp Gerontol 45: 466-472.
- Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552: 335-344.
- SabensLiedhegner EA, Gao XH, Mieyal JJ (2012) Mechanisms of altered redox regulation in neurodegenerative diseases--focus on S-glutathionylation. Antioxid Redox Signal 16: 543-566.
- Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48: 158-167.
- Lu SP, Lin Feng MH, Huang HL, Huang YC, Tsou WI, et al. (2007) Reactive oxygen species promote raft formation in T lymphocytes. Free RadicBiol Med 42: 936-944.
- Hu D, Serrano F, Oury TD, Klann E (2006) Aging-dependent alterations in synaptic plasticity and memory in mice that overexpress extracellular superoxide dismutase. J Neurosci 26: 3933-3941.
- Rao RK, Clayton LW (2002) Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem Biophys Res Commun293: 610-616.
- O’Loghlen A, Pérez-Morgado MI, Salinas M, MartÃn ME (2003) Reversible inhibition of the protein phosphatase 1 by hydrogen peroxide. Potential regulation of eIF2 alpha phosphorylation in differentiated PC12 cells. Arch Biochem Biophys 417: 194-202.
- Groeger G, Quiney C, Cotter TG (2009) Hydrogen peroxide as a cell-survival signaling molecule. Antioxid Redox Signal 11: 2655-2671.
- Lü JM, Lin PH, Yao Q, Chen C (2010) Chemical and molecular mechanisms of antioxidants: experimental approaches and model systems. J Cell Mol Med14: 840-860.
- Grune T, Reinheckel T, Davies KJ (1997) Degradation of oxidized proteins in mammalian cells. FASEB J 11: 526-534.
- Hegde ML, Hegde PM, Rao KS, Mitra S (2011) Oxidative genome damage and its repair in neurodegenerative diseases: function of transition metals as a double-edged sword. J Alzheimers Dis 24: 183-198.
- Castellani RJ, Nunomura A, Rolston RK, Moreira PI, Takeda A, et al. (2008) Sublethal RNA oxidation as a mechanism for neurodegenerative disease. Int J MolSci 9: 789-806.
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, et al. (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Bio l39: 44-84.
- Massaad CA, Klann E (2011) Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid Redox Signal14: 2013-2054.
- Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298-300.
- Harman D (1972) The biologic clock: the mitochondria? J Am Geriatr Soc 20: 145-147
- Miquel J, Economos AC, Fleming J, Johnson JE Jr (1980) Mitochondrial role in cell aging. Exp Gerontol 15: 575-591.
- Norenberg MD, Rao KV (2007) The mitochondrial permeability transition in neurologic disease. NeurochemInt 50: 983-997.
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, et al. (2005) Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308: 1909-1911.
- Popa-Wagner A, Mitran S, Sivanesan S, Chang E, Buga AM (2013) ROS and brain diseases: the good, the bad, and the ugly. Oxid Med Cell Longev 2013: 963520.
- Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J (2004) The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 62: 1984-1999.
- Su B, Wang X, Lee HG, Tabaton M, Perry G, et al. (2010) Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci Lett 468: 267-271.
- Pérez M, Cuadros R, Smith MA, Perry G, Avila J (2000) Phosphorylated, but not native, tau protein assembles following reaction with the lipid peroxidation product, 4-hydroxy-2 nonenal. FEBS Lett 486: 270-274.
- Liu Q, Smith MA, Avilá J, DeBernardis J, Kansal M, et al. (2005) Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radic Biol Med 38: 746-754.
- Misonou H, Morishima-Kawashima M, Ihara Y (2000) Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry 39: 6951-6959.
- Tillement L, Lecanu L, Papadopoulos V (2011) Alzheimer’s disease: effects of β-amyloid on mitochondria. Mitochondrion 11: 13-21.
- Mariani E, Polidori MC, Cherubini A, Mecocci P (2005) Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J Chromatogr B Analyt Technol Biomed Life Sci 827: 65-75.
- Mao P, Manczak M, Calkins MJ, Truong Q, Reddy TP, et al. (2012) Mitochondria-targeted catalase reduces abnormal APP processing, amyloid β production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum Mol Genet 21: 2973-2990.
- Obeso JA, RodrÃguez-Oroz MC, Benitez-Temino B, Blesa FJ, Guridi J, et al. (2008) Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov Disord 23: S548-S559.
- Xiang W, Schlachetzki JC, Helling S, Bussmann JC, Berlinghof M, et al. (2013) Oxidative stress-induced posttranslational modifications of alpha-synuclein: specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol Cell Neurosci 54: 71-83.
- Li J, O W, Li W, Jiang ZG, Ghanbari HA (2013) Oxidative stress and neurodegenerative disorders. Int J Mol Sci 14: 24438-24475.
- Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF (2010) Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim Biophys Acta 1802: 122-134.
- Yadav R, Chanu SI, Raj K, Sarkar S (2013) Rise and Fall of Reactive Oxygen Species (ROS): Implications in Aging and Neurodegenerative Disorders. Cell Dev Biol 2: e122.
- Olanow CW (1990) Oxidation reactions in Parkinson’s disease. Neurology 40: S32-S37.
- Andersen JK (2004) Oxidative stress in neurodegeneration: cause or consequence? Nat Med 10: S18-S25.
- Perier C, Bové J, Dehay B, Jackson-Lewis V, Rabinovitch PS, et al. (2010) Apoptosis-inducing factor deficiency sensitizes dopaminergic neurons to parkinsonianneurotoxins. Ann Neurol 68: 184-192.
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, et al. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277: 1990-1993.
- Mitomi Y, Nomura T, Kurosawa M, Nukina N, Furukawa Y (2012) Post-aggregation oxidation of mutant huntingtin controls the interactions between aggregates. J Biol Chem 287: 34764-34775.
- Patten DA, Germain M, Kelly MA, Slack RS (2010) Reactive oxygen species: stuck in the middle of neurodegeneration. J Alzheimers Dis 20: S357-S367.
- Wang JC (2010) Good cells gone bad: the cellular origins of cancer. Trends Mol Med 16: 145-151.
- Klaunig JE, Kamendulis LM, Hocevar BA (2010) Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol 38: 96-109.
- Harrison IP, Selemidis S (2014) Understanding the biology of reactive oxygen species and their link to cancer: NADPH oxidases as novel pharmacological targets. Clin Exp Pharmacol Physiol 41: 533-542.
- Nourazarian AR, Kangari P, Salmaninejad A (2014) Roles of oxidative stress in the development and progression of breast cancer. Asian Pac J Cancer Prev 15: 4745-4751.
- Sherr CJ (2004) Principles of tumor suppression. Cell 116: 235-246.
- Irwin ME, Rivera-Del Valle N, Chandra J (2013) Redox control of leukemia: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 18: 1349-1383.
- Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, et al. (1997) Mitogenicsignaling mediated by oxidants in Ras-transformed fibroblasts. Science 275: 1649-1652.
- Noda N, Wakasugi H (2001) Cancer and Oxidative Stress. JMAJ 44: 535-539.
- Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, et al. (2006) Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethylisothiocyanate. Cancer Cell 10: 241-252.
- Reddy MM, Fernandes MS, Salgia R, Levine RL, Griffin JD, et al. (2011) NADPH oxidases regulate cell growth and migration in myeloid cells transformed by oncogenic tyrosine kinases. Leukemia 25: 281-289.
- Gupta SC, Hevia D, Patchva S, Park B, Koh W, et al. (2012) Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid Redox Signal 16: 1295-1322.
- Balistreri CR, Candore G, Accardi G, Colonna-Romano G, Lio D (2013) NF-κB pathway activators as potential ageing biomarkers: targets for new therapeutic strategies. Immun Ageing 10: 24.
- Dhar A, Young MR, Colburn NH (2002) The role of AP-1, NF-kappaB and ROS/NOS in skin carcinogenesis: the JB6 model is predictive. Mol Cell Biochem 235: 185-193.
- Vurusaner B, Poli G, Basaga H (2012) Tumor suppressor genes and ROS: complex networks of interactions. Free Radic Biol Med 52: 7-18.
- Liu G, Chen X (2006) Regulation of the p53 transcriptional activity. J Cell Biochem 97: 448-458.
- Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, et al. (2005) The antioxidant function of the p53 tumor suppressor. Nat Med 11: 1306-1313.
- Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B (1997) A model for p53-induced apoptosis. Nature 389: 300-305.
- Myatt SS, Brosens JJ, Lam EW (2011) Sense and sensitivity: FOXO and ROS in cancer development and treatment. Antioxid Redox Signal 14: 675-687.
- De Ruiter ND, Burgering BM, Bos JL (2001) Regulation of the Forkhead transcription factor AFX by Ral-dependent phosphorylation of threonines 447 and 451. Mol Cell Biol 21: 8225-8235.
- Greer EL, Brunet A (2005) FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 24: 7410-7425.
- Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, et al. (2004) FOXO transcription factor activation by oxidative stress mediated by the small GTPaseRal and JNK. EMBO J 23: 4802-4812.
- Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villén J, et al. (2006) A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125: 987-1001.
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, et al. (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011-2015.
- Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, et al. (2005) Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 308: 1181-1184.
- Walczak-Jedrzejowska R, Wolski JK, Slowikowska-Hilczer J (2013) The role of oxidative stress and antioxidants in male fertility. Cent European J Urol 66: 60-67.
- Brugh VM 3rd, Lipshultz LI (2004) Male factor infertility: evaluation and management. Med Clin North Am 88: 367-385.
- MacLeod J (1943) The role of oxygen in the metabolism and motility of human spermatozoa. American Journal of Physiology 138: 512-518.
- Jones R, Mann T, Sherins R (1979) Peroxidative breakdown of phospholipids in human spermatozoa, spermicidal properties of fatty acid peroxides, and protective action of seminal plasma. Fertil Steril 31: 531-537.
- Aitken RJ, Harkiss D, Buckingham D (1993a) Relationship between iron-catalysed lipid peroxidation potential and human sperm function. J Reprod Fertil 98: 257-265.
- Aitken RJ, Harkiss D, Buckingham DW (1993b) Analysis of lipid peroxidation mechanisms in human spermatozoa. Mol Reprod Dev 35: 302-315.
- Aitken RJ (1994) The Amoroso Lecture. The human spermatozoon--a cell in crisis? J Reprod Fertil 115: 1-7.
- Fisher HM, Aitken RJ (1997) Comparative analysis of the ability of precursor germ cells and epididymal spermatozoa to generate reactive oxygen metabolites. J Exp Zool 277: 390-400.
- Plante M, de Lamirande E, Gagnon C (1994) Reactive oxygen species released by activated neutrophils, but not by deficient spermatozoa, are sufficient to affect normal sperm motility. Fertil Steril 62: 387-393.
- Fraczek M, Kurpisz M (2005) The redox system in human semen and peroxidative damage of spermatozoa. PostepyHig Med Dosw (Online) 59: 523-534.
- Agarwal A, Nallella KP, Allamaneni SS, Said TM (2004) Role of antioxidants in treatment of male infertility: an overview of the literature. Reprod Biomed Online 8: 616-627.
- Aitken RJ, Krausz C (2001) Oxidative stress, DNA damage and the Y chromosome. Reproduction 122: 497-506.
- Kodama H, Yamaguchi R, Fukuda J, Kasai H, Tanaka T (1997) Increased oxidative deoxyribonucleic acid damage in the spermatozoa of infertile male patients. Fertil Steril 68: 519-524.
- Shen H, Ong C (2000) Detection of oxidative DNA damage in human sperm and its association with sperm function and male infertility. Free Radic Biol Med 28: 529-536.
- Aitken RJ, Koopman P, Lewis SE (2004) Seeds of concern. Nature 432: 48-52.
- Showell MG, Brown J, Yazdani A, Stankiewicz MT, Hart RJ (2011) Antioxidants for male subfertility. Cochrane Database Syst Rev 1: CD007411.