Review Article
Diabetic Neuropathy: Its Pathogenesis and Therapeutic Drug Targets
Shruti Thakur and Rachana*
Corresponding author: Rachana, Department of Biotechnology, Jaypee Institute of Information Technology, A-10, Sector62, Noida, U.P- 201307, India,; E-mail: rachana.dr@iitbombay.org
Citation: Shruti T, Rachana. Diabetic Neuropathy: Its Pathogenesis and Therapeutic Drug Targets. J Cell Sci Molecul Biol. 2014;1(1): 102.
Copyright © 2014 Rachana et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Cell Science & Molecular Biology | Volume: 1, Issue: 1
Abstract
Diabetic neuropathy is the one of the most common complication which occurs in 60% of the patients suffering from diabetes. Extensive research has identified some major pathways which lead to micro-vascular complications in diabetes. Study of these pathways for the progression of the disease and their interconnections with other pathways may guide us to develop therapeutic regime for its treatment. Reactive oxygen species are reported to be one of the main important causes for diabetic neuropathy, and mitochondrial generation of reactive oxygen species in response to hyperglycemic conditions has also been considered as a contributor to oxidative stress. The increased level of oxidative stress results in the over-activation of poly ADP-ribose polymerase (PARP), as well as of NF- κB. Other than reactive oxygen species, there are many pathogenetic pathways which are involved with this complication. Some of the pathways responsible for the dreaded disease are: advanced glycation end product formation, protein kinase C, NF-κB activation and increased polyol flux. These pathways can further lead to increased cellular oxidative damage to organelles and biomolecules, causing dysregulation of cellular functions. In turn, it also leads to the production of a number of other factors which again increase the malfunction of the neurovascular system. Present article describes few important pathways responsible for diabetic neuropathy, aiming towards the proper understanding of their interlinks with other pathways, which can bring forth new drug target sites and can result in the discovery of potentially new and better therapeutics.
Keywords: Diabetic neuropathy, therapeutics, pathogenetic pathways, drug targets, oxidativestress
Introduction
Diabetes is one of the most common metabolic disorders. Lack of insulin production or resistance of the target cell towards insulin gives rise to this condition of hyperglycemia. Mostly the modern life stylelike: lack of physical activity increase in stress level (mental, physical and emotional) andchange in eating habits, playscrucial role in onset of diabetes in patients who are at risk [1]. The most morbid as well as,the most common post diabetic complication in patients with diabetes is diabetic neuropathy (DN). Patients with either type 1 or type 2 diabetes may suffer from this neurovascular disability at the time of diagnosis/later stages [2]. It occurs mostly in patients who are unable to control their blood glucose level and have sedentary lifestyle. High blood glucose concentration increases oxidative stress (OS) in the body system directly or indirectly, and it has been implicated as a main factor for the onset and progression of many post diabetic complications. Various pathways have been suggested to be linked with the progression of DN and most of themseem toinvolveincrease in oxidative stress [3] as a common point. Finding the most appropriate targets for DN for therapeutic purpose has been the main focus of the research in this area.Understanding the molecular pathways leading to DN and their interconnections, might help us to point at prospective interventions for future use. In present article, we are trying to analyze these pathways and their interconnections so as to shortlist the possible potential targets where therapeutics can be implemented to stop the progression of this morbid disease indiabetic patients.
Materials and Methods
For this review the various research and review articles were searched on Medscape, Google scholar, Pubmed with the keywords: diabetic neuropathy, oxidative stress, therapeutics, drug targets, treatments, individually and in combinations with each other.
Pathways in Diabetic Neuropathy and their Targets
Various external and internal factors may cause generation and release of Reactive Oxygen Species (ROS) increasing the oxidative stress in the biological system. Normally, system’s antioxidant level balances number of ROS species, but in prolonged stress and hyperglycemic conditions, an imbalance between the two arises [4], which in turn can lead to neurovascular and many more complications in patients. The various pathways leading to this conditionare closely linked to each other in a close circuit form. Few very important pathways are described as below:
Polyol Pathway
The hyperglycemic conditions known to activate the polyol pathway that converts excess of glucose to sorbitol by the enzyme aldose reductase. This subsequently gets converted into fructose by sorbitol dehydrogenase [5]. As glucose is now directed towards the polyol pathway, rather than the glycolytic pathway, accumulation of sorbitol and fructose takes place in the cells. Cell membrane is impermeable to sorbitol and to compensate this concentration gradient, osmolytes such as myoinositol and adenosine move out of the cells [5]. Absence of myoinositol in the cytoplasm reduces formation of ATP due to exhaustion of phosphatidylinositol [[6]] and hence the Na+/K+ - ATPase activity is reduced. This results in impaired axonal transport of nerves leading to nerve damage [7]. Also, conversion of glucose to sorbitol depletes NADPH and directly induces oxidative stress [8].
Inhibitors for polyol pathway
Aldose reductase inhibitors: The inhibitors of AR can reduce the flux of glucose to polyol pathway which in turn can reduce the accumulation of sorbitol and fructose in the cell [9]. Fidarestat, a principal Aldose Reductase Inhibitor (ARI), has been shown to have potential to improve nerve conduction and provides symptomatic relief in DN [9]. Another ARI is Ranirestat, whose application has shown improvement in motor nerve function [10]. One more example of ARI is Epalrestat that has been approved for treatment of DN. Epalrestat has shown to reduce accumulation of sorbitol in erythrocytes, sciatic nerve and ocular tissues in animals, and in erythrocytes in humans. It also improves motor and sensory nerve conduction velocity [11].
Inhibiting NADPH oxidase: NADPH oxidase is the prime mediator of oxidative stress in DN hence, if this enzyme could be inhibited, it would be a very successful therapeutic strategy for diminishing the symptoms of neuropathy. Administration of angiotensin,anNADPH oxidase isoform4 (NOX4) inhibitor, hasshown positive results in animal models and improvement seen in nerve injury and alleviation in oxidative stress [12] is reported.
Hexosamine Pathway
Similar to polyol pathway, this pathway also gets activated when there is excess glucose present in the cell. Here, the intermediate product of glycolysis, fructose-6-phosphate shiftsto enter hexosamine pathway and getsconverted to glucosamine-6-phosphate by the enzyme glutamine fructose-6-phosphate aminotransferase (GFAT) [13]. This in turn leads to the formation of uridine-diphosphate-Nacetylglucosamine, that attaches to serine and threonine residues of transcription factors, and is responsible for the increased level of expression on transcription factor Sp1 as seen in Figure1. Activated Sp1 results in overexpression of transforming growth factors (TGF-αand TGF-β) and plasminogen activator inhibitor-1 (PAI-1) that increases the complications in diabetes [14].
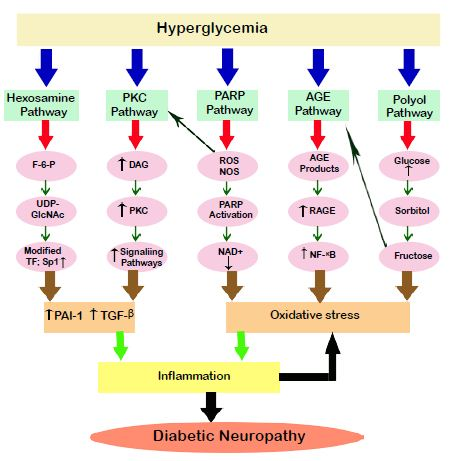
Figure 1: Pathways in diabetic neuropathy. Hyperglycemia sets forth the activation of various pathways in the target cells. AGE and polyol pathway changes the redox capacity of the cells by generation of the oxidative stress. PARP, Hexosamine and PKC leads to inflammation by increasingthe expression of transformation factors which ultimately leads to diabetic neuropathy. Inflammation and oxidative stress play a combined role in thepathogenesis of the disease. PKC: Protein kinase C, PARP: Poly(ADP) ribose phosphate, AGE: Advanced glycation end products, DAG: Diacyl glycerol, ROS: Reactive oxygen species, RNS: Reactive nitrogen species, F-6-P: Fructose 6 phosphate, UDPGlcNAc: Uridine diphosphate-Nacetylglucoseamine, RAGE: Receptor for advanced glycation end product, NAD+ Nicotinamide adenine dinucleotide, TF: Transcription factors, NF- κB: Nuclear factor κ B, PAI-1: Plasminogen activator factor-1, TGF-β:Transforming growth factor-β.
Hexosamine pathway Inhibitors
Benfotiamine,a synthetic S-acyl derivative of thiamine,decreasesthe hexosamine pathway flux and results in lowering the oxidative stress. Along with this, benfotiamine also helps in relieving pain associated with diabetic neuropathy [15]. Azaserine is also able to prevent the glucose transport to the hexosamine pathway. Moreover, GFAT inhibitor also helps in stopping the hexosamine pathway and stops the progression of neuropathy upto some extent [16].
Advanced Glycated End Products (AGEs)
Increased sugar level in blood accelerates the attachment of carbohydrate groups to proteins, lipids and nucleic acids nonenzymatically. These products undergo reactions to form advanced glycated end products (AGEs) [17]. Extracellular AGE binds to the receptor RAGE and starts the inflammatory process by employing reactive species as secondary messengers [13]. It has been found out that, the AGE-RAGE complex plays an important role in progression of diabetic neuropathy [18].
Advanced glycation end product inhibitors
Diabetic neuropathy is aggravated by the oxidative stress producedby the AGEs and its subsequent activation of AGE receptor. Inhibitors for AGE and RAGE can be forthcoming therapeutic approaches for tackling DN. Benfotiamine; the hexosamine pathway inhibitor is a good antioxidant and has anti-AGE formation properties [19]. Additionally, thiazolium compounds like ALT-711 (algebrium), C36, TRC4186, and TRC4149 and their prototype N-phenacylthiazolium bromide (PTB) are also known to stop the formation of AGE [20]. They work to break the pre-accumulated AGE products or break the existing AGE cross-links.
Other than the above examples of RAGE inhibitors, lowmolecular- weight heparin (LMWH) can bind to RAGE and act as antagonist to RAGE [211]. LMWH treatment in animal models have shown encouraging effects for treating albuminuria and resulted in increased glomerular cell number and mesangial expansion in a dose-dependent manner [21]. Thiazolidinediones, calcium channel blockers, angiotensin-converting enzyme inhibitors (ACEIs), angiotensin II receptor blockers (ARBs), and statins also help suppress RAGE expression [22].
Protein Kinase C (PKC) Pathway
The Protein Kinase C is one of the important factors in proper functioning of the nerves and the progression of DN. PKC is a family of 11 isoforms, out of which 9 are linked with the production of oxidative stress [23]. Hyperglycemia stimulates the formation of Di-acyl glycerol (DAG) which activates these 9 isoforms [24]. These isoforms lead to stimulation of the expression of signalling pathways involving PAI-1 (Plasminogen Activator Inhibitor-1), NF-κB and TGF-β, also depicted in Figure 1 [25]. They then lead to overproduction ofcytokines and induce inflammatory response. It affects the nerves in terms of their contractibility and permeability [26]. Also, it leads to inhibition of Na+/K+ ATPase. PKC also actuatesstress genes, phosphorylating transcription factors, affects the balance of gene expression and induces oxidative stress [14].
Inhibitors of Protein Kinase C pathway
Activation of PKC pathway produces free radicals leading to diabetic microvascular complications. Studies propose that hyperglycemia leads to the activation of PKCβ, which is considered to have potential role in microvascular complications of DN [27]. Hyperglycemic activation of PKCβ causes abnormal signalling and other complications like cytokine activation and inhibition, vascular alterations, cell cycle and transcriptional factor deregulation, and abnormal angiogenesis [28]. Ruboxistaurin, which inhibits PKC-β activation, has been particularly successful in curbing the progress of DN, but is pending to get approval [29].
On the other hand, PKCδ plays a major role in islet cell function and insulin response. The changes in the level and activity of PKCδ in mice strains correlates with risk of glucose tolerance and insulin resistance. Along with it, loss of inflammation is also associated with insulin resistance in PKCδ deficient mice. Therefore, inhibition of PKCδ may also offer a treatment for metabolic syndrome [30]. One more target of PKC pathway is VEGF (glycoprotein) which is activated due to PKC pathway and leads to retinopathy. Pegaptanib sodium is an anti-VEGF aptamer which binds to VEGF and prevents it from interacting with its receptors [31].Results show that pegaptanib improved visual acuity outcomes, and preserved central retinal thickness [32]. Two more examples for VEGF inhibitor are Ranibizumab and bevacizumab [33]. They are the recombinant monoclonal antibody fragment and full-length antibody, respectively. Both of them are derived from the same mouse monoclonal antibody precursor and have high affinity for VEGF. Many case studies have recommended bevacizumab to be effective in decreasing retinalneovascularization and macular edema in diabetics. Studies show ranibizumab to be superior in improving visual acuity and regression of neovascularization [34].
PARP Pathway
Poly ADP-ribose polymerase is family of proteins which act as nuclear enzymes and are involved in number of processes out of which DNA repair and programmed cell death are the important ones. Generally, cells suffering from PARP deficiency are prone to more DNA damage than the cells having PARP. However, overactivationof PARP is also harmful and is seen in diabetic patients. During hyperglycemia, the high glucose in blood interacts with the endothelium and induces the release of mediators of electron transport chain. Polymers of ADP-ribose and nicotinamide areactivated by utilizing NAD+. As NAD+ is used up by PARP pathway, other pathways which use NAD+ as a cofactor are compromised. These compromised metabolic pathways result in functional loss and irregularities like oxidative stress and cell death increasing the morbidity in diabetic neuropathy.
PARP Pathway Inhibitors
As discussed above, PARP activation is involved in the pathogenesis of DN, and its inhibition lightens numerous experimental pathologic conditions connected with oxidative stress in DN. Inhibiting PARP is of great importance as the compromised pathways will be able to function properly and glucose will not beshunted to alternate pathways like AGE and PKC. Nicotinamide has been shown to act as a PARP inhibitor and an antioxidant in animals that improves complications of early DN [14].
Inflammation
Increase in oxidative stress and many other pathways leads to inflammation which is found to be responsible for most of the diabetic complications [35,36]. Two of the inflammatory agents is, C-reactive protein and TNF-α(Tumour necrosis factor-α). These factors are responsible for the proinflammatory response in the nerve tissues that presentthe symptoms and develop neuropathy [37]. As mentioned earlier, initiators of inflammatory factors such as NF-κB, TGF-αand TGF-β are produced from various pathways during hyperglycemia [25]. There is a two way relation between ROS and inflammation meaning ROS leads to inflammation and inflammation further adds up to increase in ROS. Cyclooxygenase-2 is anenzymewhich is upregulated by NF- κB generating prostaglandin E2 and further produces ROS [38]. An additional inflammatory enzyme, which also catalysed by NF-κB is iNOS (inducible nitric oxide synthetase) [14]. NF-κB is the central point of activation and is the centre of inflammatory response in hyperglycemic conditions. Along with it, NF-κB induces the production of cytokines in endothelial cells, Schwann cells and neurons and absorption of macrophages also occurs in diabetic nerves. Presence of excessive number of macrophages impairs regeneration of nerves in DN and advertently leads to increased production of cytokines and ROS [39].
Additionally, nuclear factor- Kappa (NF-k) light chain enhancer of B cell or NF-κB is a transcription factor responsible for the production of proinflammatory cytokines. On the other hand, the nuclear factor-2 erythroid related factor- (Nrf2) is a redox regulated transcription factor involved in the regulation of antioxidantdefence systems [40]. In healthy cells and tissues, Nrf2 and NF-κB are regulated to maintained redox homeostasis. During pathological conditions like in diabetic neuropathy, the control slips in the form of over-activation of NF-κB and simultaneous suppression of Nrf2. Although Nrf2 is briefly activated by oxidative stress, extracellular related kinase (ERK) activation restrains permanent Nrf2 activation [41]. Waning in Nrf2 activity and an insistent increase in NF- κB activity can lead to enhanced nitrosative and oxidative stress. These further results in collective damage to peripheral nerve fibers, reduced blood supply to neuronal tissue, release of prostaglandins, causing hypersensitivity to pain and hence, lead to the development of neuropathic pain [42].
Anti inflammatory therapeutics
As, inflammation is one of the main causes of diabetic neuropathy complications, controlling inflammation is thereby on central importance in curbing the neuropathic disorder. In this line of research, one of the molecule Erythropoietin’s anti-inflammatory effects has been shown to help in protection of blood flow andconduction velocities in sciatic and sural nerves [43]. Trandolapril has also shown significant reduction in neuropathic complication by inhibiting angiotensin, which is a powerful vasoconstrictor along with possessing pro-inflammatory properties [40]. Also, selective inhibition of cyclooxygenase-2 enzyme has been shown to reduceautonomic neuropathy in animal models, supporting the view of promising therapeutic benefits of anti-inflammatory agents [44].
Few more examples of anti-inflammatory molecule are curcumin, melatonin, resveratrol and sulphoraphane. Studies have shown their beneficial effects in suppressing increased activity of NF-κB. Also, treatment with these agents increased the levels of Nrf2 which inhibits the TNF-αdependent activation of NF-κB in endothelial cells [45].In case of TNF-αα, its increased expression is found in the kidney of the patients suffering from the DN. Also the level of TNF- αis more in patients having microalbuminuria. Pentoxifylline inhibits the expression of mRNA levels of TNF-α[46]. Also, adiponectin derived from adipose tissue suppresses the inflammatory marker including the TNF- α[47]. The administration of insulin and antioxidant therapy has been reported to reduce TNF- αand improvement in neuropathy. Drugs like angiotensin converting enzyme inhibitors and beta blockers have shown to have anti TNF properties [12].
Problems associated with Treatment of DiabeticNeuropathy
Despite much knowledge about the pathogenesis of diabetic neuropathy and its pathways, not much improvement has occurred in terms of its cure. Moreover, although most of the pathways have been extensively researched, there is a wide margin of unknown, which is yet to be discovered. Thus, poorly understood mechanisms also create a hurdle in effective treatment of the disorder. One major cause forthe unsuccessful cure for neuropathy is irreversible nerve damage and so, even if the major events responsible for DN disappear, the nerve cannot be regenerated. There have been instances of several drugs being declined in clinical trials because of intolerable side effects while some are yet to be in clinical trials which might have great impact in curing diabetic neuropathy at some point in future. Even though extensive research is going on and many fruitful results from preclinical studies have been reported, but whether these will be beneficial in therapeutics is still a matter of debate. Recently, scientists are exploring stem cell based therapies for DN and other diseases and positive results have been reported in related areas like treating diabetic foot. One study concluded the a graft composed of Mesenchymal Stem Cells taken from the patient’s bone marrow and combined with fibroblasts on collagen membrane (Coladerm) helped in healing of diabetic foot ulcers. The results showed decreased wound size and increased vascularity through this combined therapy [48]. We expect that stem cell therapy would solve the problem associated with regeneration of nerves in this case also and might be successful if combined with the above mentioned therapeutic regimes.
Conclusion
Treatment of diabetic neuropathy is contained by strict glycemic control and balanced lifestyle but, is not shown to be sufficient to treat it. Although, many drugs targeting metabolic sites in various pathways have been identified and researched upon, crucial advances are still needed for better targets aiming for reversal of diabetic neuropathy. Many antioxidants (natural and synthetic) have also been reported to have positive effects as they might overcome the oxidative stress and delay the progression of pathogenesis.
Although, clinical trials have reached final stages for some drugs while some have been approved in specific countries, their worldwide acceptance is still a hope in future. Only two drugs till date have been approved for use: Epalrestat in Japan and α-Lipoic acid in Germany. The FDA in U.S.A. does not allow its use for neuropathy patients as their clinical trials are still inconclusive. Aleglitazar is still undergoing phase III clinical trials whereas Ruboxistaurin has shown effect intreating retinopathy but is not effective against neuropathy.
The future aspects in the field of the treatment of diabetic neuropathy can be seen in the advent of therapeutic targets at mitochondrial metabolic control level and inflammatory pathways. They are most likely to diminish the incidence of diabetic neuropathy. Stem cell based therapies are also claiming attention due to their promising role in rectifying this debilitating condition.
Acknowledgment
We are thankful to department of Biotechnology, JIIT Noida for providing the requisite support for this study.
References
- Young MJ, Boulton AJ, MacLeod AF, Williams DR, Sonksen PH (1993) A multicentre study of the prevalence of diabetic peripheral neuropathy in the United Kingdom hospital clinic population. Diabetologia 36: 150–154.
- Veves A, Backonja M, Malik RA (2008) Painful diabetic neuropathy: epidemiology, natural history, early diagnosis, and treatment options. Pain Med 9: 660–674.
- Banba N, Nakamura T, Matsumura M, Kuroda H, Hattori Y, et al. (2000) Possible relationship of monocyte chemoattractant protein-1with diabetic nephropathy. Kidney Int 58: 684–690.
- Rahman K (2007) Studies on free radicals, antioxidants, and co-factors. Clin Interv Aging 2: 219-236.
- Evans JL, Goldfine ID, Maddux BA, Grodsky GM (2002) Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 23: 599–622. .
- Busui R, Herman W, Feldman E, Low PA, Martin CL, et al. (2010) DCCT and EDIC studies in type 1 diabetes: lessons for diabetic neuropathy regarding metabolic memory and natural history. Curr Diab Rep 10: 276–282.
- Brownlee M (2005) The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54: 1615–1625.
- Athans W, Stephens H (2008) Open calcaneal fractures in diabetic patients with neuropathy: a report of three cases and literature review. Foot Ankle Int 29: 1049–1053.
- Hotta N, Toyota T, Matsuoka K, Shigeta Y, Kikkawa R (2001) Clinical efficacy of fidarestat, a novel aldose reductase inhibitor, for diabetic peripheral neuropathy: a 52-week multicenter placebo controlled double-blind parallel group study. Diabetes Care 24: 1776–1782.
- Bril V, Hirose T, Tomioka S, Buchanan R (2009) Ranirestat for the management of diabetic sensorimotor polyneuropathy. Diabetes Care 32: 1256–1260.
- Vincent AM, Callaghan BC, Smith AL, Feldman EL (2011) Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat Rev Neurol 7: 573-583.
- Benter IF, Yousif MH, Dhaunsi GS, Kaur J, Chappell MC, et al. (2008) Angiotensin‑(1–7) prevents activation of NADPH oxidase and renal vascular dysfunction in diabetic hypertensive rats. Am J Nephrol 28: 25–33.
- Jain SP (2004) Callaghan BC, Cheng HT, Stables CL, Smith AL, Feldman EL (2012) Diabetic neuropathy: clinical manifestations and current treatments. Lancet Neurol 11: 521– 534.
- Edwards JL, Vincent AM, Cheng HT, Feldman EL (2008) Diabetic neuropathy: mechanisms to management. Pharmacol Ther 120: 1–34.
- Haupt E, Ledermann H, Kopcke W (2005) Benfotiamine in the treatment of diabetic polyneuropathy—a three-week randomized, controlled pilot study (BEDIP study). Int J Clin Pharmacol Ther 43: 71–77.
- Filipps A, Clark S, Proietto J (1997) Increased flux through the hexosamine biosynthesis pathway inhibits glucose transport acutely by activation of protein kinase C. Biochem J 324: 981–985.
- Toth C, Rong LL, Yang C, Martinez J, Song F, et al. (2008) Receptor for advanced glycation end products (RAGEs) and experimental diabetic neuropathy. Diabetes 57: 1002–1017.
- Mahmood D, Singh BK, Akhtar M (2009) Diabetic neuropathy: therapies on the horizon. J Pharm Pharmacol 61: 1137–1145.
- Shakher J, Stevens MJ (2011) Update on the management of diabetic polyneuropathies, Diabetes Metab Syndr Obes 4: 289–305.
- Schwedler SW, Verbeke P, Bakala H, Weiss MF, Vilar J, et al. (2001) N-phenacylthiazolium bromide decreases renal and increases urinary advanced glycation end products excretion without ameliorating diabetic nephropathy in C57BL/6 mice. Diabetes Obes Metab 3: 230–239.
- Myint KM, Yamamoto Y, Doi T, Kato I, Harashima I, et al. (2006) RAGE control of diabetic nephropathy in a mouse model: effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes 55: 2510–2522.
- Marx N, Walcher D, Ivanova N, Rautzenberg K, Jung A, et al. (2004) Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes 53: 2662–2668.
- Geraldes P, King GL (2010) Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res 106: 1319–31.
- Cameron NE, Cotter MA (2002) Effects of protein kinase C beta inhibition on neurovascular dysfunction in diabetic rats: interaction with oxidative stress and essential fatty acid dysmetabolism. Diabetes Metab Res Rev 18: 315–323.
- Cotter MA, Jack AM, Cameron NE (2002) Effects of the protein kinase C beta inhibitor LY333531 on neural and vascular function in rats with streptozotocin-induced diabetes. Clin Sci (Lond) 103: 311–321.
- Rajbhandari SM, Piya MK (2005) A brief review on the pathogenesis of human diabetic neuropathy: observations and postulations. International J Diabetes and Metabolism 13: 135–140.
- Meier M, Menne J, Park JK,Haller H (2007) Nailing down PKC isoform specificity in diabetic nephropathy. Nephrol Dial Transplant 22: 2421-2425.
- Cameron NE, Cotter MA (2002) Effects of protein kinase C beta inhibition on neurovascular dysfunction in diabetic rats: interaction with oxidative stress and essential fatty acid dysmetabolism. Diabetes Metab Res Rev 18: 315–323.
- Vinik AI, Bril V, Kempler P, Litchy WJ, Tesfaye S, et al. (2005) Treatment of symptomatic diabetic peripheral neuropathy with the protein kinase C ð›½- inhibitor ruboxistaurinmesylate during a 1-year, randomized 15 placebo-controlled, double-blind clinical trial. Clin Ther 27: 1164–1180.
- Mima A, Kitada M, Geraldes P, Matsumoto M, Mizutani K, et al. (2012) Glomerular VEGF resistance induced by PKC delta/SHP-1 activation and contribution to diabetic nephropathy. The FASEB J 26: 2963–2974.
- Soulis T, Thallas V, Youssef S, Gilbert RE, McWilliam BG, et al. (1997) Advanced glycation end products and their receptors co-localise in rat organs susceptible to diabetic microvascular injury. Diabetologia 40: 619–628.
- Cumbie BC, Hermayer KL (2007) Current concepts in targeted therapies for the pathophysiology of diabetic microvascular complications. Vasc Health Risk Manag 3: 823–832.
- Simo R, Hernandez C (2008) Intravitreous anti-VEGF for diabetic retinopathy: hopes and fears for a new therapeutic strategy. Diabetologia 51: 1574-1580.
- Papadopoulos N, Martin J, Wiegand SJ (2012) Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 15: 171-185.
- Negi G, Kumar A, Joshi RP, Ruby PK, Sharma SS (2011) Oxidative stress and diabetic neuropathy: current status of antioxidants. The IIOABJ 2: 71-78.
- Obrosova IG, Drel VR, Pacher P, et al. (2005) Oxidative-nitrosative stress and poly (ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation isrevisited. Diabetes 54: 3435–3441.
- Yagihashi S, Mizukami H, Sugimoto K (2011) Mechanism of diabetic neuropathy: where are we now and where to go? Journal of Diabetes Investigation 2: 18–32.
- Busui RP, Marinescu V, Huysen VC (2002) Dissection of metabolic, vascular, and nerve conduction interrelationships in experimental diabetic neuropathy by cyclooxygenase inhibition and acetyl-L-carnitine administration. Diabetes 51: 2619–2628.
- Bierhaus A, Haslbeck KM, Humpert PM (2004) Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J Clin Invest 114: 1741–1751.
- Hosseini A, Abdollahi M, (2013) Diabetic Neuropathy and Oxidative Stress: Therapeutic Perspectives. Oxidative Med Cell Longev 168039.
- Zheng H, Whitman SA, Wu W (2011) Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 60: 3055–3066.
- Palsamy P, Subramanian S (2011) Resveratrol protects diabetic kidney by attenuating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via Nrf2-Keap1 signaling, Biochim Biophys Acta, 7: 719–731.
- Loesch A, Tang H, Cotter MA, Cameron NE (2010) Sciatic nerve of diabetic rat treated with epoetin delta: effects on C‑fibers and blood vessels including pericytes. Angiology 61: 651–668.
- Kellogg AP, Wiggin TD, Larkin DD, Hayes JM, Stevens MJ, et al. (2007) Protective Effects of Cyclooxygenase-2 Gene Inactivation against Peripheral Nerve Dysfunction and Intraepidermal Nerve Fiber Loss in Experimental Diabetes. Diabetes 56: 2997-3005.
- Rajasekaran A, Sivagnanam G, Xavier R (2008) Nutraceuticals as therapeutic agents: A Review. Research J Pharm Tech 1.
- Han J, Thompson P, Beutler B (1990) Dexamethasone and pen toxifylline inhibit endotoxin-induced cachectin/tumor necrosis factor synthesis at separate points in the signaling pathway. J Exp Med 172: 391–394.
- Ouchi N, Walsh K (2008) A Novel Role for Adiponectin in the Regulation of Inflammation. Arterioscler Thromb, Vasc Biol 28: 1219-1222.
- Vojtassak J , Danisovic L , Kubes M, Bakos D, Jarabek L, et al. (2006) Autologous biograft and mesenchymal stem cells in treatment of the diabetic foot. Neuro endocrinol Lett 27: 134–137.