Case Report
Neurological Variability in Acute Intermittent Porphyria: Case Reports to Highlight the Focus on Different Neurological Manifestations
Mishra N* and Raut T
Department of Neurology, Kokilaben Dhirubhai Ambani Hospital, Mumbai, Maharashtra, India
*Corresponding author:Neha Mishra, Department of Neurology, Kokilaben Dhirubhai Ambani Hospital, Mumbai, Maharashtra, India. E-mail Id: Nehamdoc94@gmail.com
Article Information:Submission: 11/02/2025; Accepted: 10/03/2025; Published: 14/03/2025
Copyright: © 2025 Mishra N, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Acute intermittent porphyria (AIP) is a genetic condition due to deficiency of porphobilinogen deaminase enzyme in the heme synthesis pathway. It has an autosomal dominant inheritance. Its manifestation includes abdominal pain, peripheral neuropathy, autonomic symptoms and renal involvement.[1]We report similar cases of a young female presenting as pure motor quadriparesis and another man with seizure and posterior reversible encephalopathy syndrome.
A 17-year-old female presented with severe intermittent abdominal pain, vomiting, followed by muscle weakness and thinning of all four limbs. She underwent various investigations before AIP was suspected. High levels of urine porphobilinogen and nerve conduction study suggestive of pure motor neuropathy were identified. Therefore, AIP was the possible diagnosis. She had a partial recovery; her clinical course of the attack episode lasted for 8 weeks.
Another 18-year-old man came with severe abdominal pain and vomiting 4 days following anterior cruciate ligament repair of right knee. He underwent endoscopy for the same and ended up with a diagnosis of erosive gastritis. A week later he developed an episode of generalised tonic clonic seizure and neuroimaging showed PRESS. He was detected to have heterozygous mutation in hydroxy-methyl bilane synthase gene, thus confirming the diagnosis of porphyria.
A 17-year-old female presented with severe intermittent abdominal pain, vomiting, followed by muscle weakness and thinning of all four limbs. She underwent various investigations before AIP was suspected. High levels of urine porphobilinogen and nerve conduction study suggestive of pure motor neuropathy were identified. Therefore, AIP was the possible diagnosis. She had a partial recovery; her clinical course of the attack episode lasted for 8 weeks.
Another 18-year-old man came with severe abdominal pain and vomiting 4 days following anterior cruciate ligament repair of right knee. He underwent endoscopy for the same and ended up with a diagnosis of erosive gastritis. A week later he developed an episode of generalised tonic clonic seizure and neuroimaging showed PRESS. He was detected to have heterozygous mutation in hydroxy-methyl bilane synthase gene, thus confirming the diagnosis of porphyria.
Keywords:Porphyria; Neuropathy; Press
Introduction
Porphyria has two major phenotypes: cutaneous and hepatic.
The estimated prevalence is around 1 in 5500 to 5800 people in
western world. Acute intermittent porphyria (AIP) is more common
and due to deficiency of the porphobilinogen deaminase enzyme
that converts porphobilinogen to hydroxymethylbilane, resulting
in accumulation of porphobilinogen and aminolevulinic acid. The
classic symptoms are severe unexplained abdominal pain along
with nausea, vomiting, or constipation; neurological attacks, such
as epilepsy, sensori-motor weakness; psychiatric symptoms, such as
agitation, depression, insomnia, psychosis, delusions, hallucinations;
autonomic disturbances, such as hypertension and tachycardia; and
hyponatremia. These symptoms are triggered by sleep deprivation,
stress, fasting, infections, some medications, and menstruation.
The wide range of penetrance (1-38%) raises concerns about the
underdiagnosis of AIP and can lead to fatal complications. [2-4]
Hence the aim is to review the varied presentation of porphyria.
Case 1
A 17-year female came with progressive weakness and thinning
of limbs for 3 months which started in upper limb (distal followed
by proximal weakness). A week later she had proximal lower
limb weakness and by the end of 1 month she was bedbound. The
weakness was maximum by the end of 6 weeks. She was given
intravenous immunoglobulinat nearby health centerafter a diagnosis
of possible acute inflammatory polyradiculopathy was made. The
nerve conduction study done at that time showed pure motor
axonal > demyelinating type of neuropathy. She did not improve
with the treatment but noted progressive thinning in all four limbs
subsequently. She presented to us after 3 months and we found that
she had acute onset progressive, upper limb onset, painless, pure
motor, are flexic disabling lower motor neuron type of quadriparesis
(Upper limb – distal > proximal, Lower limb distal and proximal)
without truncal or respiratory involvement, sensory, autonomic,
cranial nerve or sphincter involvement.
The patient underwent several investigations before AIP was
suspected. High levels of urine porphobilinogen and a follow up
nerve conduction study suggestive of pure motor neuropathy
were identified. Therefore, AIP was the possible diagnosis. She
had a partial recovery; her episode lasted 8 weeks. She has been
advised carbohydrate rich diet and asked to avoid the list of trigger
medications.
Our case is similar to one presented by Mutluay et al where a
17-year-old female presented with rapidly progressive quadriparesis
with autonomic dysfunction, initially suspected to be an acute motor
axonal neuropathy variant of Guillain-Barre syndrome but later
found to be due to porphyria.[5]
Case 2
18-year-old man presented with severe abdominal pain and
vomiting 4 days following anterior cruciate ligament repair, which
did not resolve with symptomatic medications. His imaging studies

including ultrasonography and CT abdomen was unremarkable
thus he underwent endoscopy and ended up with a diagnosis of
erosive gastritis. A week later he developed an episode of generalised
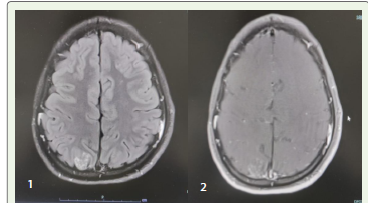
tonic clonic seizure and neuroimaging/ MRI Brain showed bilateral
posterior predominant T2/FLAIR hyperintensities s/o PRESS.
(MRI brain showed cortical edema with gyral swelling and altered
signal changes of both frontal and right parietal and both occipital
lobes along with clustered nodular enhancement of right pre cuneus
with sulcal effacement s/o PRESS)
His mother also had similar h/o convulsion following similar
episode of abdominal discomfort we suspected him to have porphyria.
Surgery was the stressful event which precipitated the attack. Any
surgery is a stressful procedure which when combined with factors
like dehydration, fasting or exposure to certain drugs or anesthesia
can increase the body”s demand for heme, leading to buildup of
porphyria precursors which can trigger porphyria attack.[6] His urine
for delta ALA was positive and subsequently he was detected to have
heterozygous mutation in HMBS gene, thus confirming the diagnosis
of porphyria. We managed him with dextrose containing intravenous
fluids and symptomatic management with which he recovered well.
Both the patients were advised to have carbohydrate rich diet and list
of unsafe medications were explained.
Our case is similar to one published by Andrew et al wherein
PRESS was found to be one of the complications in patients with
porphyria. [7]
Discussion
Hemeconsists of 64 kDa tetrameric structure. Heme is present in
hemoglobin, myoglobin, respiratory cytochromes, and cytochrome
P450 enzymes. Heme is formed from glycine and succinyl coenzyme
which consists of 8 enzymatic steps, four enzymes are present in the
cytosol, and four enzymes are present in the mitochondria. Hepatic
ALA synthase1 enzyme is the rate limiting enzyme which is inhibited
by heme. [[7,8,2, 3, 9, 10.]
Neuropathy occurs in 10-40 % of porphyria cases and is due
to neurotoxicity caused by accumulated porphyrin precursors
and dysfunction of Na/K ATPase pump leading to abnormal
axon transport and neural dysfunction. The neuropathy is usually
pure motor, axonal, proximal upper limb onset and symmetric.
Distal paresthesias are less common. Cranial nerve involvement is
infrequent.[11]
Seizures are seen in approximately 10-20% patients with
symptomatic porphyrias. The most common seizure types are tonicclonic
or complex partial seizures. Status epilepticus is less common.
The possible mechanism may be due to neurotoxic substance
presumably ALA/PBG which may interact with GABA or glutamate
receptors. Endothelial dysfunction, hypoperfusion, vasoconstriction
in the setting of neurotoxicity leads to compromise of blood brain
barrier and brain edema. PRES may be a rare manifestation due to the
same mechanism. Other neurological symptoms include autonomic
dysfunction, encephalopathy, coma, agitation, anxiety, depression,
insomnia and hallucinations.[1]
Precipitating factors for Acute Porphyria
• Drugs—barbiturates, oestrogens, methyldopa, danazol, diazepam, phenytoin, carbamazepine, sulphonylureas sulphonamides, chloramphenicol, tetracyclines, some antihistamines
• Fasting
• Smoking
• Surgery
• Alcohol
• Substance particularly marijuana, cocaine, ecstasy and amphetamines
• Infection
• Premenstrual attacks are common
• Drugs—barbiturates, oestrogens, methyldopa, danazol, diazepam, phenytoin, carbamazepine, sulphonylureas sulphonamides, chloramphenicol, tetracyclines, some antihistamines
• Fasting
• Smoking
• Surgery
• Alcohol
• Substance particularly marijuana, cocaine, ecstasy and amphetamines
• Infection
• Premenstrual attacks are common
Diagnosis:
Patients with suspected signs and symptoms suggestive of
AIP should be asked about potential triggers and family history of
porphyria. A publicly available database of porphyrinogenic drugs
enlists the causative agents. (https://porphyriafoundation.org/
drugdatabase/) laboratory investigation may reveal hyponatremia,
leukocytosis, mild transaminitis, red or brown urine (not related
to hemoglobin or bilirubin) without any evidence of infectious,
gastrointestinal, hepatobiliary, pancreatic, renal, or gynecologic
cause. Mild to severe hyponatremia in seen in 25-60% of cases.[12]During acute attacks, the urinary porphyrin precursor
Porphobilinogen (PBG) is usually increased in AIP. The Trace PBG
Kit detects elevated levels of urinary PBG. ALA and PBG levels
are often elevated. Also, urinary (and fecal) porphyrin analyses
can suggest a specific acute hepatic porphyria. Once a biochemical
diagnosis is ensured, genetic mutation analysis for AIP should be
undertaken. Molecular diagnostic studies also are to be done for
confirming diagnosis of patients with symptoms, to identify at-risk
family membersand to offer asymptomatic heterozygote counseling
to avoid the drugs, fasting, hormones, and other precipitanting
factors.[10]
Treatment:
Initial management focuses on eliminating factors such as
medications, caloric deprivation, and dehydration that may be
precipitating factor. Rehydration using IV normal saline, glucose
infusions, and discontinuation of any suspected inducer medications
is a vital part of the management of the acute attack. Pain relief
using opioids is considered safe. Definitive treatment is done
with administration of IV hemin for 3-14 days, which reverses the
increase in ALAS1. Research studies are being conducted with small
interfering RNA (siRNA) to ALAS1 and appear promising. Givosiran
was approved by the FDA in November 2019 for the treatment of
acute hepatic porphyrias in adults.Ongoing management: some patients may have recurring
attacks. This includes females during menstruation. Gonadotropin releasing
hormone analogs have been found useful to prevent ovulation in female patients that present with recurrent premenstruation related acute porphyria.
Acute seizure management can be a challenging in acute
porphyrias as most anticonvulsants induce the cytochrome P450
enzyme. Acute seizure management includes the following:
• First-line medication: Magnesium sulfate and diazepam
• For status epilepticus: Lorazepam, per rectal diazepam
• Correction of metabolic risk factors: Such as correction of hyponatremia with normal saline considering the volume status of the patient.
• Long-term seizure control: Gabapentin
• First-line medication: Magnesium sulfate and diazepam
• For status epilepticus: Lorazepam, per rectal diazepam
• Correction of metabolic risk factors: Such as correction of hyponatremia with normal saline considering the volume status of the patient.
• Long-term seizure control: Gabapentin
Patients might have autonomic dysfunction, which can be
managed by beta-blockers. An acute rise in blood pressure can be
treated with appropriate emergency medication, such as labetalol.
Psychiatric symptoms are managed by giving phenothiazines,
such as chlorpromazine.
Neuromuscular symptoms are treated with Gabapentin, early rehabilitation, mechanical ventilation for respiratory weakness and nasogastric feeds for severe dysphagia. Acute attacks of neuropathic pain can be managed with hemin infusion and opioids. In most cases recovery is often incomplete.[10,11]
Neuromuscular symptoms are treated with Gabapentin, early rehabilitation, mechanical ventilation for respiratory weakness and nasogastric feeds for severe dysphagia. Acute attacks of neuropathic pain can be managed with hemin infusion and opioids. In most cases recovery is often incomplete.[10,11]
Statement and Disclosure:
No person who had contributed substantially to the production
of this manuscript had been excluded from authorship. Person who
has contributed partially have been acknowledged in the manuscript.Nothing to disclose (financial or non-financial interests) that are directly or indirectly related to the work submitted for publication.
References
Citation
Mishra N, Raut T. Neurological Variability in Acute Intermittent Porphyria: Case Reports to Highlight the Focus on Different Neurological Manifestations. Indian J Neurol. 2025;6(1): 143.