Case Report
A Rare Case of an Atypical Friedreich’s Ataxia
Sanjo K John, N Shobana, M Sacratis, CJ Selvakumar and V Sadeesh Kumar
Department of Neurology, Coimbatore Medical College, Tamil Nadu, India
*Corresponding author:Sanjo K John, Department of Neurology, Coimbatore Medical College, Trichy Road, Coimbatore, Tamil Nadu, India. E-mail Id: sanjokalathil24@gmail.com
Article Information:Submission: 16/09/2024; Accepted: 07/10/2024; Published: 10/10/2024
Copyright: © 2024 John SK, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Friedreich’s ataxia is one of the most common causes of young
onset ataxia. It is a degenerative ataxia initially described by Nicolaus
Friedreich, as a clinical syndrome characterized by gait ataxia,
incoordination, peripheral neuropathy, absent reflexes, abnormalities
in eyes movement, pes cavus, scoliosis, cardiomyopathy, and diabetes
mellitus.[1] It is an autosomal recessive disorder, with disabilities
usually starts from second decade. Patients suffering from this disease
dies at early age due to cardiomyopathy and heart failure. Friedreich’s
ataxia is due to GAA repeats in long arm of chromosome 9 that
affects frataxin gene.[2] Harding, in 1981,described typical form of
Friedreich’s ataxia with essential criteria such as onset of symptoms
before 25 years of age, progressive ataxia of limbs and gait, absent
knee and ankle jerks, reduced motor wave conductions, extensor
plantar reflex, and onset of dysarthria after five years of disease
onset.[3] Additional criteria included are pyramidal signs in both
limbs, scoliosis, hammer toe, pes cavus, absent reflexes in upper
limbs, and abnormal ECG.[2] The MRI reports in the early stages may
not demonstrate obvious cerebellar atrophy, but shows cervical spinal
cord atrophy. Late stage of the disease will have marked cerebellar
atrophy in MRI scans. Studies have reported red flags for excluding
the possibility of a diagnosis of Friedreich ataxia, which include
prominent cerebellar atrophy, preserved sensory action potential,
and mental retardation.[4]
Literature has arbitrarily subdivided Friedreich ataxia based on
the delayed age of onset into late-onset (25-39 years) and very late onset
Friedreich ataxia (>40 years).[5] Late-onset cases found to
have atypical features including preserved deep tendon reflexes or
spasticity with hyperreflexia.[6] Atypical phenotypes of Friedreich’s
ataxia also have been described in the literature. Schulz et al (2009)
listed out the recessive ataxias with similar characteristic features
such as ataxia with vitamin E deficiency, ataxia with oculomotor
apraxia, and autosomal-recessive spastic ataxia of Charlevoic-
Saguenay, which can be considered for differential diagnosis.[7] Here
we describe an atypical phenotype presentation of Friedreich’s ataxia
with retained reflexes.
Case Report
A 34-year-old male, born of second-degree consanguineous
marriage, presented with history of unsteadiness in walking noticed at
the age of 16 years in the form of swaying to both sides while walking.
The symptoms had insidious onset and was gradually progressing. Five
years later, patient had sensory symptoms in the form of decreased
pain and temperature perception in the lower limbs. The patient also
reported to have history suggesting of wash-basin symptoms and
double vision. At the same time, patient was diagnosed with diabetes
mellitus and was given insulin injection. Further medical evaluation
also detected the presence of cardiomyopathy. Gradually, patient
had difficulty in speech in the form of articulating words syllableby-
syllable. Patient also had stiffness of both lower limbs, which
progressed to an extent that lower limbs were very stiff with scissoring
of legs, and he was not able to stand or walk without support. He was
almost bedbound after one year, even requires assistance for sitting
on bed. The family history revealed similar pattern of symptoms for
his paternal uncle.
On evaluation, knuckle pigmentation, hammer toe, pes cavus and
scoliosis were identified on general examination. The higher mental
functions were found to be normal. Cranial nerve examination revealed
nystagmus and square wave jerks. The muscle tone was increased
in both lower limbs. However, the ankle jerks were absent while all
other tendon reflexes were present. The plantar reflexes had extensor
response bilaterally, suggesting involvement of corticospinal tract.
Pain, touch, vibration, and position sense were impaired bilaterally.
Further evaluation identified bilateral impairment in finger-to-finger
and finger-to-nose test, and the presence of dysdiadochokinesia and
rebound phenomenon, suggestive of cerebellar involvement. There
was cerebellar scanning speech, nystagmus, and square wave jerks.
Tandem gait could not be elicited as patient could not stand or walk.
On routine investigation, random blood sugars were high. The
ECG recordings showed T-inversion in anterior and inferior leads.
The ECHO test results reflected hypertrophic cardiomyopathy. The
nerve conduction study revealed bilateral upper limb and lower limb
axonal neuropathy. Ophthalmological and ENT examinations were
unremarkable.
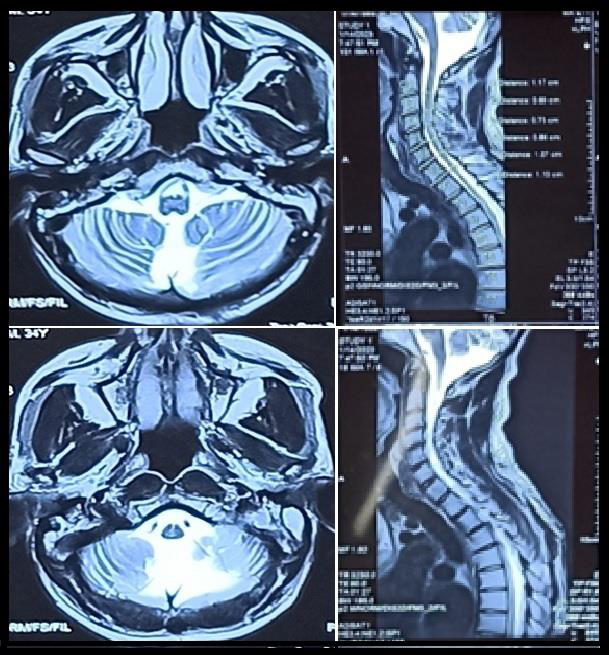
MRI Brain scans showed cerebellar atrophic changes and cervical
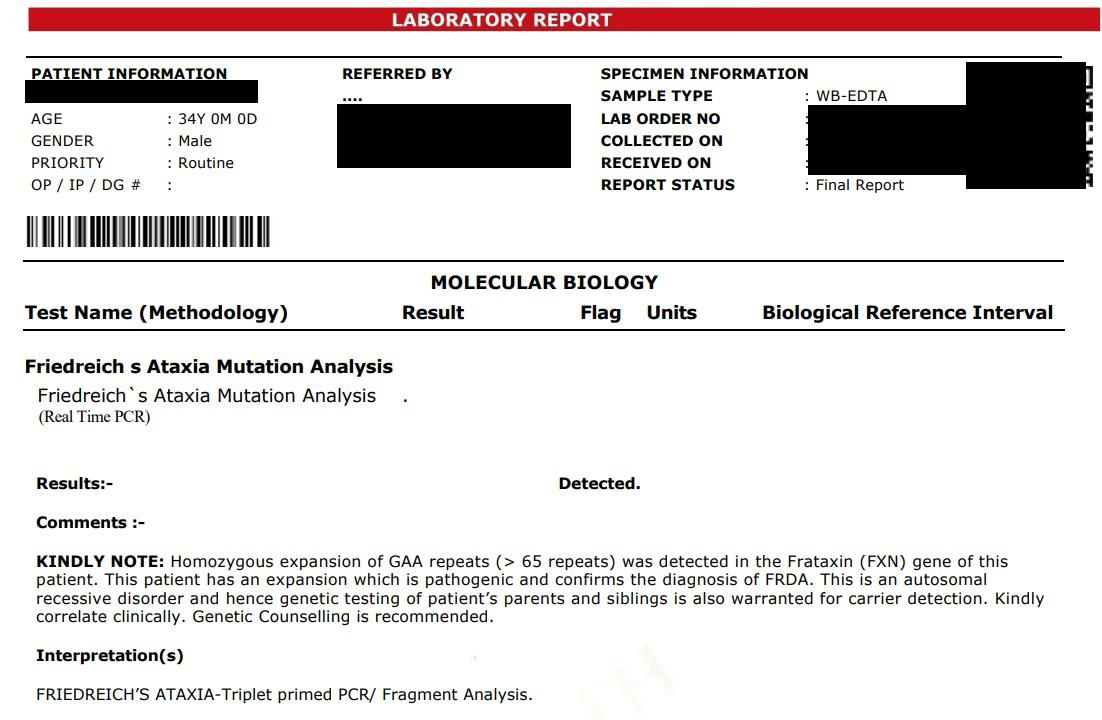
spinal canal narrowing [Figure 1]. Friedreich’s ataxia mutational
analysis was done for the patient by real time PCR method which
revealed homozygous expansion of GAA repeats (> 65 repeats) in the
frataxin gene [Figure 2]. Thus, patient had a definitive diagnosis of
atypical Friedreich’s ataxia, with preserved reflexes.
Patient was treated with insulin to control blood sugar and
diazepam to reduce spasticity. In view of cardiomyopathy with severe
LV dysfunction, furosemide and spironolactone were prescribed after
cardiologist opinion. Patient also received in-patient physiotherapy,
and was discharged at request after 1 week.
Discussion
Friedreich’s ataxia is considered as the most common autosomal
recessive ataxia. [8] The neurological features reported in typical
form Friedreich’s ataxia include gait ataxia, limb ataxia, weakness and
wasting which is prominent in the lower limbs, areflexia, sensory loss,
abnormalities of the eye movements, dysarthria, dysphagia, auditory
neuropathy, and sphincter disturbances.[2] Late stages of the disease
reported to have marked cerebellar atrophy.
Atypical phenotypes of Friedreich’s ataxia as reported in the
literature are Late Onset Friedreich’s ataxia (LOFA), Very Late
Onset Friedreich’s Ataxia (VLOFA), and Friedreich’s ataxia with
retained reflexes.[9]The reported patient also had severe spasticity,
and retained deep tendon reflexes, suggesting atypical phonotype of
Friedreich’s ataxia. Klockgether et al., reported 30-40% variance in
the presence of spasticity in atypical phenotypes.[10] Literature has
also reported atypical variants with cervical cord atrophy,[2] which
was also present in this patient.
While studies have reported lower incidence of non-neurological
features such as skeletal deformities and cardiac involvement in
atypical phenotypes,[11] scoliosis, pes cavus, cardiomyopathy and
diabetes were reported in our patient. Cardiomyopathy is reported
to be absent in the atypical phenotypes,[2] but the reported patient
had hypertrophic cardiomyopathy with abnormal T-wave inversion.
Delatycki (2009) reported the progression of Friedreich ataxia to be
slow, with a mean time of 36 years between onset and death.[12]
Studies have reported treatment strategies with coenzyme Q, vitamin
E, Idebenone, and L-carnitine as medical options, which enhances
mitochondrial function and act as free radical scavangers.[7]
Our patient presented here had core clinical features of
Friedreich’s ataxia which was genetically confirmed, and exhibited
severe spasticity and retained tendon reflexes, suggestive of an atypical
phenotype of Friedreich’s ataxia. Knowledge of atypical presentations
of Friedreich’s ataxia is very important in clinical practice. It will help
us differentiate Friedreich’s ataxia from other differential diagnoses.
References
Citation
John SK, Shobana N, Sacratis M, Selvakumar CJ, Sadeesh Kumar V. A Rare Case of an Atypical Friedreich’s Ataxia. Indian J Neurol. 2024;5(1): 134.