Case Report
Osmotic Demyelination Syndrome As a Manifestation of Hypokalemia Secondary to Sjogren’s Syndrome With Renal Tubular Acidosis
Sonali S, Manali C* and Rohidas B
Department of General Medicine, BJGMC & SGH, Pune
*Corresponding author: Manali C, Department of General Medicine, BJGMC & SGH, Pune; Tel: +91 9757353019; E-mail:
chaudharimanali96@gmail.com
Article Information: Submission: 15/10/2022; Accepted: 12/11/2022; Published: 14/11/2022
Copyright: © 2022 Sonali S, et al. This is an open access article distributed under the Creative Commons Attribution
License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is
properly cited.
Abstract
Sjogren’s syndrome is a female dominated autoimmune disorder with diverse phenotypical expression. For most patients, disease runs an indolent
course with sicca symptoms, musculoarticular pain and disabling fatigue. Distal real tubular acidosis is an extra-glandular manifestation of Sjogren’s
syndrome characterized by failure of kidney to secret hydrogen ions. This results in increased secretion of other cations including potassium with resultant
metabolic acidosis and hypokalemia.
We present herewith a case of Sjogren’s syndrome with distal renal tubular acidosis who presented with a wide spectrum of neurological manifestations.
Further evaluation of this patient along with the brain imaging revealed osmotic demyelination. As patient did not have hyponatremia at any point during
hospitalization, we could attribute occurrence of Osmotic demyelination syndrome to hypokalemia only.
Introduction
Sjogren’s syndrome is a chronic, slowly progressing autoimmune
disease characterized by lymphocytic infiltration of the exocrine
glands resulting in xerostomia and dry eyes (keratoconjunctivitis
sicca). Extraglandular manifestations are seen in one third of patients
with Sjogren’s syndrome. Renal involvement in Sjogren’s syndrome
is not uncommon and may precede sicca symptoms. The pathology
in most of the cases is tubulointerstitial nephritis causing distal renal
tubular acidosis [1-5]. It indicates failure of the intercalated cells in
the collecting duct of kidney to secrete hydrogen ions. As a result,
secretion of other cations, including potassium, is increased to
maintain electroneutrality. Potassium loss can result in hypokalemic
paralysis, often presenting as recurrent episodes. However, cerebellar and extra-pyramidal manifestations are uncommon due to primary
neurological involvement in Sjogren’s syndrome [6,7].
Case History
A 43 years old female, residing in a slum area, a homemaker,
presented with chief complaints of loose motions since two days,
vomiting since one day and weakness in both lower limbs since eight
to ten hours.
On examination, the patient was afebrile, had a pulse rate of 60
beats/min and a blood pressure of 90/60 MM of mercury. No signs of
dehydration were present.
Her cardiovascular, respiratory and abdominal system
examinations were normal.
On CNS examination, patient was drowsy but arousable. Her
speech was slurred with stress on some syllables. Cranial nerve
examination was normal. Motor system examination revealed
power of grade five in both upper limbs and grade one in both
lower limbs. Her deep tendon reflexes were absent in all four limbs.
She had an intention tremor and finger nose test was positive on
cerebellar examination. Sensory examination was within normal
limits. Bilateral plantar responses were flexor. On the next day, she
developed weakness in both upper limbs as well with a power of grade
three (3/5).
Her lab report showed Serum Sodium- 136.4mEq/L(normal
range-:125-145mEq/L), Serum Potassium- 1.19mEqu/L(normal
range-:3.5-5.5mEq/L). She received potassium correction in the form
of infusion of injection potassium chloride in normal saline. After
24 hours of correction patient’s muscle weakness and sensorium
improved significantly but serum potassium remained low. Samples
for urinary electrolytes and serum magnesium were sent which
revealed normal serum magnesium and a positive urine anion
gap. ABG revealed PH - 7.388(normal range-:7.35-7.45), Chloride-
135 mEq/L(normal range-:102-119mEq/L), Potassium- 1.4mEq/
L(normal range-:3.5-5.5mEq/L). The picture was suggestive of distal
renal tubular acidosis. Owing to the persistence of cerebellar signs
we ordered an MRI of brain with the possibility of CNS vasculitis.
MRI revealed features of osmotic demyelination syndrome with
extrapontine myelinolysis.
Other lab parameters were as follows-:
Her hemogram showed Hemoglobin of 11.2g/dl(11-18g/dl), Total leucocyte Count-8140/uL(4000-11,000/uL), platelet count-
1,21,000/uL(1,20,000-5,00,000/uL).
Liver function tests were within normal limits. Ultrasound
examination of abdomen was normal.
ECG - Widening of QRS complex, RBBB, ST segment depression
with ‘T’ wave flattening in leads V1 to V6 and generalized U waves.
Taking into consideration, a picture of distal Renal tubular
acidosis and MRI suggestive of osmotic demyelination syndrome,
we contemplated Sjogren’s syndrome to be the culprit and got ANA
Blot assayed which revealed SM/RNP-58AU (normal range 6-12AU),
SSA/RO60KD-75AU (normal range 6-12AU), SS/RO52KD-
69AU (normal range 6-12AU). Patient had no evidence of other
rheumatological disorders. The patient satisfied 4 out of 6 criteria
for Sjogren’s syndrome as per the American-European Consensus
criteria and thus, the diagnosis of primary Sjogren’s syndrome was
made. She was started on oral potassium supplements and Shohl’s
solution.
Around 6 months ago, she had presented with subacute diarrhea
with hypokalemia that responded to symptomatic treatment. The
episode was self-limited.
Discussion
We had a case of 43 years old female presenting with acute
diarrhea. During the second episode, features of hypokalemia
dominated the clinical picture. Presence of additional neurological
signs led us to the diagnosis of osmotic demyelination syndrome.
On evaluation of etiology, the hypokalemia occurred as a result of
distal renal tubular acidosis secondary to Sjogren’s syndrome. She
had pyramidal with extra pyramidal features on the day of admission
itself. These findings were explained by the MRI picture of Osmotic
demyelination syndrome involving pontine as well as extra pontine
areas. The quadriparesis was secondary to hypokalemia.
We attribute the Osmotic demyelination syndrome findings to
hypokalemia only as the patient did not have hyponatremia at any
point during hospitalization. Also, the findings were demonstrated
before her hypokalemia was corrected, obviating any potential role
of Intravenous fluids administered for correction of hypokalemia.
Several case reports have been documented in literature regarding
hypokalemia as one of the implicated mechanisms in the causation
of Osmotic demyelination syndrome. M D Norenberg hypothesized
that hyponatremia caused cerebral endothelial injury by osmotic
dysregulation, further leading to release of myelinotoxic factors
causing vasogenic cerebral edema [1]. A similar mechanism could
have resulted in Osmotic demyelination syndrome associated with
hypokalemia Figure 1 & 2.
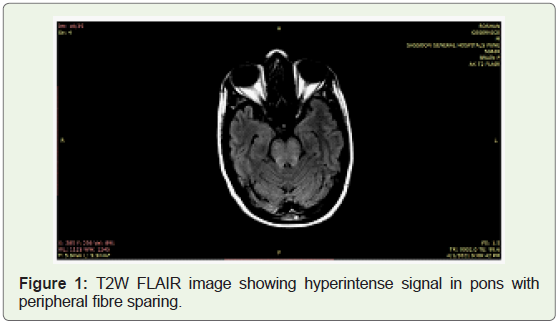
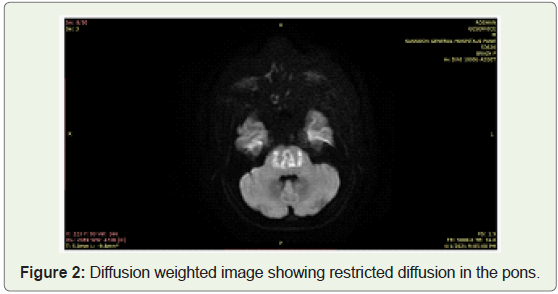
I Dørup et al have documented the correlation between cellular
potassium: sodium ratio and concentration of H-ouabain binding
sites. Potassium deficiency possibly leads to downregulation of
sodium-potassium pumps in skeletal muscles [2]. A mechanism
close to this finding could be operational in endothelial or glial
cell membranes of the central nervous system as well. Decreased
concentration of Na-K ATPase in endothelial cell membrane during
hypokalemia may predispose the cell susceptible to injury by slight increase in osmotic stress [3]. Therefore, there is a possibility that
Osmotic Demyelination syndrome was induced by a slight increase in
osmotic pressure attributable to fluid infusion such as of electrolytes
in the presence of severe hypokalemia, even if the increase rate was so
low as to not induce injury in normal state.
Thus, the findings of Sjogren’s syndrome with distal Renal tubular
acidosis, manifesting as hypokalemia and Osmotic demyelination
syndrome, are of a rare occurrence.
This lady showed resolution of symptoms gradually as her
potassium levels were normalised. After day 10 of admission, her
serum potassium was 3.7mEq/L and there was no focal neurological
deficit on examination. On follow up visit, she had no signs of
hypokalemia and was asked to continue oral potassium supplements.
In our case, there was a conspicuous absence of hyponatremia. Thus,
hypokalemia leading to downregulation of sodium potassium pumps
in glial cell membranes of the central nervous system could be the
possible mechanism. The cerebellar dysfunction improved after the
correction of hypokalemia. The patient followed up with us after one
month and on examination showed no residual neurological deficit.
Thus, Hyponatremia is not the only implicating factor in the
causation of osmotic demyelination syndrome. Hypokalemia should
be corrected and maintained in normal reference range depending
on the cause.
Acknowledgement
We thank Dr Shefali Pawar, Professor and Head, Dept of
Radiology for her expert opinion on radiological imaging of this
patient.
References
Citation
Sonali S, Manali C*, Rohidas B. Osmotic Demyelination Syndrome As a Manifestation of Hypokalemia Secondary to Sjogren’s Syndrome With Renal Tubular Acidosis. Indian J Neurol. 2022;3(1): 113.