Case Report
GNE Myopathy- A Rare Distal Myopathy with Rimmed Vacuoles: A Case Report
Agrawal V1, Chavali P2, Lagishetty V1, Kachu R1 and Gongati NC1*
1Department of Neurology, Yashoda Hospitals, Secunderabad, India
1Neuropathologist, Yashoda Hospitals, Secunderabad, India
*Corresponding author: Gongati NC, Department of Neurology, Yashoda Hospitals, Secunderabad, India; Phone - +91
8919589498, E-mail: nissichrysoliteg5@gmail.com
Article Information: Submission: 16/08/2022; Accepted: 09/09/2022; Published: 12/09/2022
Copyright: © 2022 Agrawal V, et al. This is an open access article distributed under the Creative Commons
Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the
original work is properly cited.
Abstract
GNE myopathy is a rare distal myopathy also known as hereditary inclusion body myopathy (HIBM) or Nonaka disease or Distal Myopathy with Rimmed
Vacuoles (DMRV) or IBM2. It’s an autosomal recessive disease that can present in homozygous or compound heterozygous forms, with predominantly
compound heterozygous mutations found in Indian subcontinent. Clinically most striking feature is distal leg weakness sparing quadriceps, distinguishing it
with other common myopathies. GNE myopathy most commonly presents in the third decade of life as foot drop because of predominant Tibialis Anterior
muscle involvement. Later it progresses to other muscles of lower and upper limbs; however Quadriceps usually remain spared even in advanced stages.
Muscle biopsy typically shows rimmed vacuoles with atrophied fibres and congophilic depositions. Distal muscle weakness with sparing of quadriceps and
rimmed vacuoles in muscle biopsy make this rare myopathy a unique one. Here we are presenting a typical case of GNE myopathy with founder mutation
(p.Val727Met) which is common in Indian population.
Keywords
GNE myopathy, Nonaka, Distal myopathy, Rimmed vacuoles
Introduction
GNE myopathy, also known as HIBM, Nonaka myopathy, IBM2
and distal myopathy with rimmed vacuoles, is a genetic disorder
that affects primarily the skeletal muscles. First signs of the disease
appear between 20 and 40 years of age and affect males and females at
the same rate. This condition is characterized by progressive muscle
weakness which typically worsens over time, decreased grip strength
and frequent loss of balance [1,2].
GNE myopathy is caused by mutations in the GNE gene, which
encodes for an enzyme known as glucosamine (UDP-N-acetyl)-2-
epimerase/N-acetylmannosamine kinase. Mutations in the GNE
gene have been reported worldwide in approximately 4,000 patients
although the incidence of the disease has been estimated to be
1-9/100,000 individuals.
GNE myopathy is a unique myopathy with distinct clinical
and pathological features so awareness of this myopathy may help
in identifying more cases in the future and thus increasing the
understanding about this myopathy. We believe that this a rare case
and that it can contribute to our knowledge about such kind of rare
diseases.
Case Report
Here we present a case of young man, 25 years born out of a
non-consanguineous marriage and second child in the family. He
presented to us with progressive difficulty in walking for duration of
one year. Initially it started with the left leg and over few months it
progressed to right leg also. He was not having any family member
suffering from similar complaints. On examination there was atrophy
in the anterior compartment of both the legs with bilateral foot drop. Other muscles in lower limb were normal including quadriceps.
There was mild weakness of bilateral hand grip which went unnoticed
by the patient. He had normal cognitive functions, cranial nerves and
sensory examination but diminished reflexes.
His blood chemistry was normal except high CPK levels
(approximately 5 times of the higher normal range). Electromyography
showed small, polyphasic motor unit potentials with early and
complete recruitment, most prominent in tibialis anterior muscles
bilaterally. Cardiac evaluation including ECG and 2-D echo was
within normal limits. Muscle biopsy from tibialis anterior showed
myofibers of variable sizes, endomysial fibrosis with fatty infiltration
and few fibres showed vacuoles with basophilic, granular material
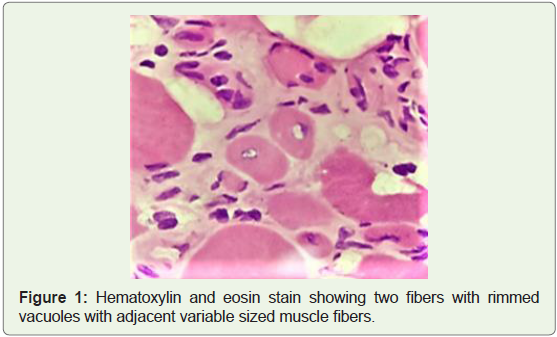
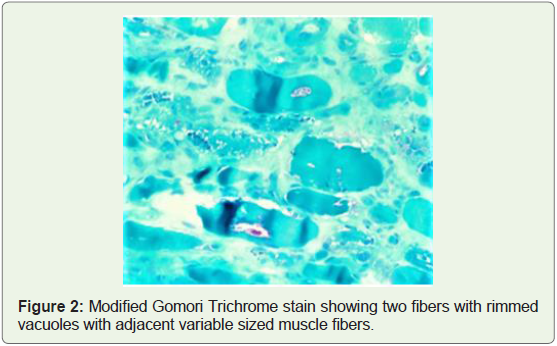
within rimmed vacuoles (Figure 1 and 2) suggestive of rimmed
vacuolar myopathy.
Whole genome analysis showed a heterozygous (amino acid
change: p.(Val727Met) Missense mutation) likely pathogenic variant
in GNE gene which is consistent with most commonly found mutation
in Indian subcontinent, in autosomal recessive GNE myopathy. The
GNE variant c.2179G>A p.(Val727Met) causes an amino acid change
from Val to Met at position 727.
Discussion
GNE myopathy was first described by Ikuya Nonaka in 1981 as a
distal myopathy with Rimmed vacuoles and lamellar (myeloid) body
depositions [3], hence it was called as Nonaka myopathy initially.
Later it was described as “quadriceps sparing myopathy” by Argov
Zohar in 1984 [4].
Clinically GNE myopathy most commonly presents in the third
decade of life as distal weakness in both legs because of predominant
Tibialis Anterior involvement. Later it progresses to proximal muscles
of lower limbs and the upper limbs with remarkable sparing of
quadriceps even in late stages. Bilateral foot drop and other muscle
involvements of lower limbs with relative sparing of quadriceps
is a good clinical sign to have a suspicion of GNE myopathy [5].
The reported patient had progressive difficulty in walking and on
examination; there was atrophy in the anterior compartment of
both the legs with bilateral foot drop. Other muscles in lower limbs
and quadriceps were normal but bilateral handgrip was weak. In
approximately 5% of patients quadriceps may be involved in early
stage making the diagnosis difficult [6]. Usually it takes 10 years for
the patient to become wheelchair bound but some patients may be
ambulatory even after 15-20 years of disease onset [7]. Disease may
have a more benign course in patients having homozygous kinase
mutation.
Cardiac involvement is not common in GNE myopathy, however
few patients are being reported to have cardiac involvement [8].
These patients typically have mild to moderately elevated CPK levels
with mild elevation of ALT sometimes [9]. In the reported patient,
cardiac evaluation was within normal limits. MRI of skeleton muscles
can help to diagnose the disease at early stage. Selective quadriceps
sparing especially vastus lateralis even in advanced stages of disease
may also be useful in diagnosis [10]. Needle Electromyography
shows myopathic pattern while spontaneous activity is not a classical
feature of GNE myopathy. Our patient Electromyography showed
small, polyphasic motor unit potentials, most prominently in tibialis
anterior muscles bilaterally.
Muscle biopsy is characterised by rimmed vacuoles, atrophied
fibres and deposition of congophilic material in vacuolated or nonvacuolated
muscle fibres. Inflammatory markers may be found in
early stage of disease and can’t help excluding hereditary inclusion
body myopathy [11]. Being a distal myopathy, biopsy is usually taken
from distal muscles including Gastrocnemius and Tibialis anterior.
Figure 1 and 2 depict muscle biopsy from tibialis anterior which
showed myofibers of variable sizes, endomysial fibrosis with fatty
infiltration and few fibres showed vacuoles with basophilic, granular
material within rimmed vacuoles suggestive of rimmed vacuolar
myopathy.
GNE myopathy is an autosomal recessive disorder that can present
in homozygous or compound heterozygous forms. Homozygous
forms are common worldwide while heterozygosity is dominant
feature in Indian subcontinent. The reported patient’s Whole
genome analysis showed a heterozygous variant (amino acid change:
p.(Val727Met) Missense mutation). Bhattacharya et al analysed 67
GNE myopathy patients from Indian subcontinent, of whom 21%
were homozygous for GNE variants, while the rest were found to
be compound heterozygous [12]. They found 35 different mutations
in the GNE gene, out of which p.Val727Met (65%) was the most
common mutation found mainly in heterozygous form. Large case
series with patients from all over India have been described by Nalini
A. et al [13,14]. p.Val727 Met variant is found at high frequency in
normal Indian population (1%-2%) and very high frequency (14%) in the normal Gujarati population. It is strongly believed that impaired
sialylation is the main cause of disease pathology [15]. The GNE
gene encodes the bifunctional enzyme, UDP- N- acetylglucosamine
2- epimerase/ N- acetylmannosamine- kinase (GNE/MNK) that
catalyses the rate- limiting step of the 5- N- acetylneuraminic acid
(sialic acid) biosynthesis [16]. Sialic acid (SA) is a modified sugar that
helps in formation of large variety of glycoproteins and glycolipids.
Conclusion
GNE myopathy is a differential diagnosis of other distal myopathies
or CMT. Currently, there is no cure for the disease and treatment
is focused on managing the symptoms. However, preclinical and
clinical studies of several potential therapies are underway, including
substrate replacement and gene therapy-based strategies.
References
Citation
Agrawal V, Chavali P, Lagishetty V, Kachu R, Gongati NC. GNE Myopathy- A Rare Distal Myopathy with Rimmed Vacuoles: A Case Report. Indian J Neurol. 2022;3(1): 112.