Case Report
A Rare Case of Movement Disorder with Developmental Delay [Clinical Phenotype of de novo Gnao1 mutation]: Case Report and Review of Literature
Satish S*, Robert Wilson, Arunan S and Kalpana
Department of Neurology, SRM Medical College and Research Center, SRM Nagar, Potheri, Chengalpattu, Tamilnadu, India
*Corresponding author: Shanmugasundaram Satish, Department of Neurology, SRM Medical College and Research
Center, SRM Nagar, Potheri, Chengalpattu, Tamilnadu, India; Phone +91 8056353093; E-mail: sundaramk60@gmail.com
Article Information: Submission: 08/03/2022; Accepted: 09/04/2022; Published: 11/04/2022
Copyright: © 2022 Satish S, et al. This is an open access article distributed under the Creative Commons Attribution
License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is
properly cited.
Abstract
Mutations in GNAO1 (guanine nucleotide-binding protein, alpha-activating activity polypeptide O) were recently identified as being causative for early
epileptic encephalopathy. Since then approximately 27 patients with severe developmental delay and different neurological phenotypes for epilepsy and
involuntary movement disorder have been reported. Here we report a 7 year old female with mutations in GNAO1 harboring the de novo mutation (c.736G >
A, p.Glu246Lys) but showing differences in phenotype with pronounced hyperkinetic movements and global developmental delay. The mutation was found
using a targeted next generation sequencing gene panel and demonstrates targeted sequencing as a powerful tool for identifying mutations in genes where
only a few de novo mutations have been identified.
Keywords
Early infantile epileptic encephalopathy; Othahara syndrome; GNAO1 mutation; Movement disorders; c.736G > A, p.Glu246Lys
Introduction
G-proteins with all their subtypes have long been recognized
to be essential for delivery of extracellular information and stimuli
via membrane-bound receptors and intracellular second messenger
systems. Heterotrimeric G-proteins consist of α-, β-, and γ-subunits.
In mammals, four Gα subtypes are known so far. GNAO1 (MIM
139311) on chromosome 16q13 encodes for Gα0-a subtype which is
known to be predominantly expressed in brain tissue [1]. On a cellular
level Gα0 works as an inhibitor of voltage-gated Ca2þ channels and
activator of inward potassium channels. Knockout of Gα0 in mice
leads to a complex and early lethal phenotype with manifestations
including tremor, severe epilepsy, and abnormal behavior [2].
Mutations in GNAO1 were first identified to be causative
for epileptic encephalopathy in four female individuals with
Ohtahara syndrome (OS) by Nakamura et al in 2013 [3]. Since
then approximately 23 additional patients have been reported with
a variable phenotype which appears to involve severe intellectual
disability and motor developmental delay in all patients, a varying
degree of either early epileptic syndromes, such as OS in some
patients and a variety of involuntary movement disorders in others
[4-14]. Until very recently only female patients had been reported
to show epileptic phenotypes whereas reported male patients were
found to show the same degree of psychomotor retardation but also
severe dystonia, which was ameliorated by deepbrain
stimulation in
some cases [5]. However, severe movement disorders also affect some of the reported female patients. A single male patient with mutations
in GNAO1 was reported only recently [6].
Here we report a 7 yr old patient with mutations in GNAO1
harboring the de novo mutation (c.736G > A, p.Glu246Lys) showing
differences in phenotype with pronounced hyperkinetic movements
and global developmental delay as compared to already reported
cases having similar mutation.
Case Report
The 7 yr old female child born out of third degree of consanguineous
marriage with a birth weight of 2.31 kg was born through LSCS at 36
weeks of gestation. At 20 weeks of intrauterine life anomaly scan done
was found to be normal. The baby cried immediately after birth. The
neonatal period was complicated by poor feeding and jaundice on
day 3 of life for which the baby warranted 1 week of NICU admission.
In addition to this, there were unspecified stereotypic jerks in bilateral
lower limbs. The baby developed social smile at 2 months of age. Head
control independent sitting was not achieved and subsequently, other
motor functions were delayed. At 3 yrs of age the patient’s mother
noted involuntary jerky movements involving bilateral upper and
lower limbs which were brief without loss of consciousness. The baby
also had dystonic posturing of the trunk and extensor spasms along
with exaggerated startle and sleep myoclonus since 4 years of age.
There were no seizure episodes in the past. At 7 years of age baby had
a fever for one week along with worsening of pre existing symptoms
along with the first episode of focal tonic seizures with extensor
spasm. The patient was developmentally delayed with no eye contact.
She was opistotonic and had severe spasticity in bilateral upper and
lower limbs. Babinski reflex was bilaterally positive. She had subtle
dysmorphic features like dolichocephalic head and high arched
palate. Her head circumference was 43 cms at the present admission.
The younger sibling of this patient had similar complaints of
involuntary movements associated with seizures for which the baby
wasn’t evaluated and died at the age of 6 yrs. No other members in the
family had a similar illness or epilepsy.
Electroencephalography was done at the 7 years of age. The
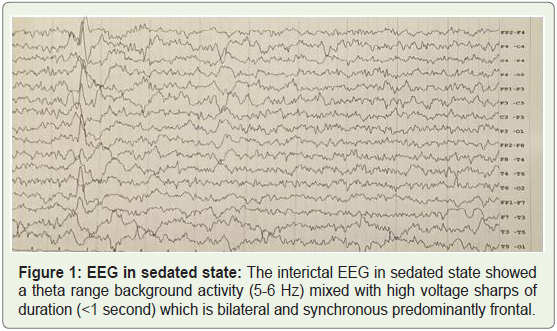
interictal EEG in sedated state showed a theta range background
activity (5-6 Hz) mixed with high voltage sharps and spikes which
is bilateral and asynchronous predominantly frontal (Figure 1). At
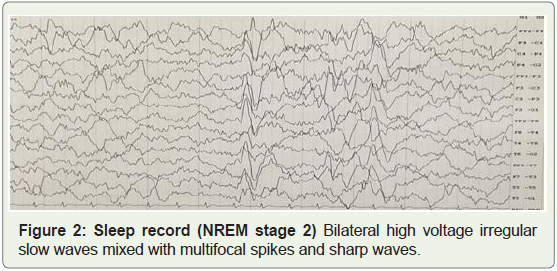
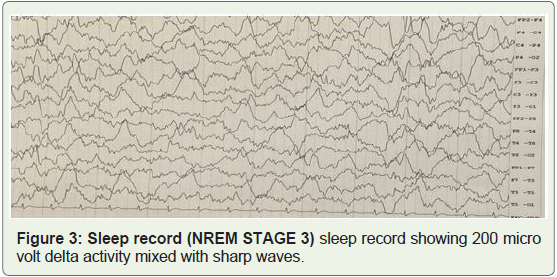
sleep, there were bilateral high voltage irregular slow waves mixed
with epileptiform activity (Figure 2&3).
MRI brain done at 9 months of age was normal. Repeat imaging
done at 4 yrs revealed mild frontotemporal atrophy with a generally
decreased amount of white matter and size of basal ganglia with some
increase in signal intensity in external capsule and periventricular
white matter on T2-weighted images.
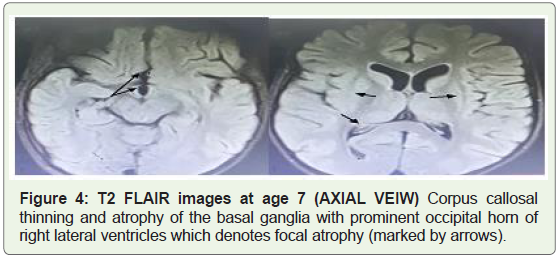
MRI brain imaging done at the age of 7 revealed bilateral basal
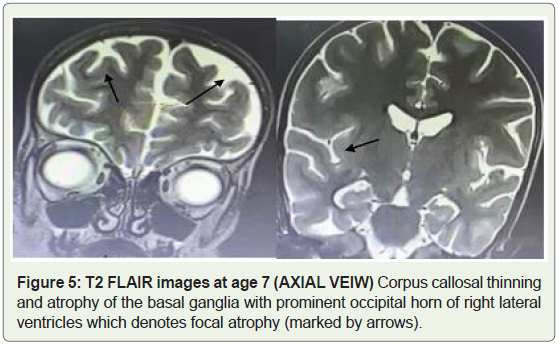
ganglia atrophy along with corpus callosal atrophy (Figure 4). T2
weighted images revealed widening of the subarachnoid spaces with
prominent sulcus (Figure 5).
During the course of the disease, the patient had multiple
extrapyramidal symptoms like myoclonic jerks, extensor spasms
and dystonic movements. She had been treated with multiple combinations of drugs including antiepileptics without many
benefits. Psychomotor development has been delayed from birth with
a gradual progression of symptoms.
During the last admission to our hospital, the baby had one
episode of hypoglycemia with metabolic acidosis, arterial pH of 7.14
and Bicarbonate values were 10 mEq/L, which subsequently got
corrected with the treatment of underlying fever. CSF analysis done
during that time was normal.
Over the years extensive metabolic work up was performed
which were all under normal limits. Work up for Inborn Errors of
Metabolism (TMS) was also found to be normal. Serum Lactate was
under normal limits (53mg/dl). Previous CSF analysis for the done
autoimmune panel was negative. At the age of 7 yrs whole exon
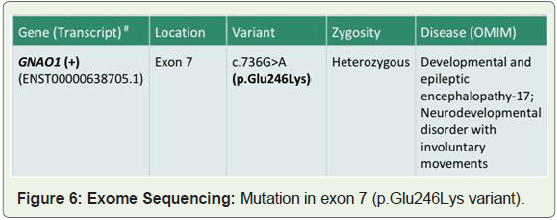
sequencing was done after obtaining consent from the parents. The
genetic exon sequencing revealed GNAO1(+) transcript at exon 7 (
variant 736 G>A ) (Figure 6).
Methods
Molecular Analyses:
We collected the DNA samples from the girl and her parents.
Genomic DNA from the family was extracted from EDTA
anticoagulated blood samples using standard methods. To identify
the disease-causing gene, targeted next generation sequencing of 40
different genes associated with childhood epilepsy was performed.
Whole exon sequencing (WES) was performed in our patient.
Identified GNAO1 mutations were validated and segregation tested
in our patient by Sanger sequencing.Results
WES was initiated and showed heterozygous de novo c.736G > A,
p.Glu246Lys mutation in exon 7 of GNAO1 in our patient but not in
the parents. The mutation was predicted to be “probably damaging”
(PolyPhen2) and “disease causing” (MutationTaster). This mutation
affects specifically exon 7 of transcript variant 1 (NM_020988.2) and
affects a highly conserved glutamic acid at position 246. Up to the
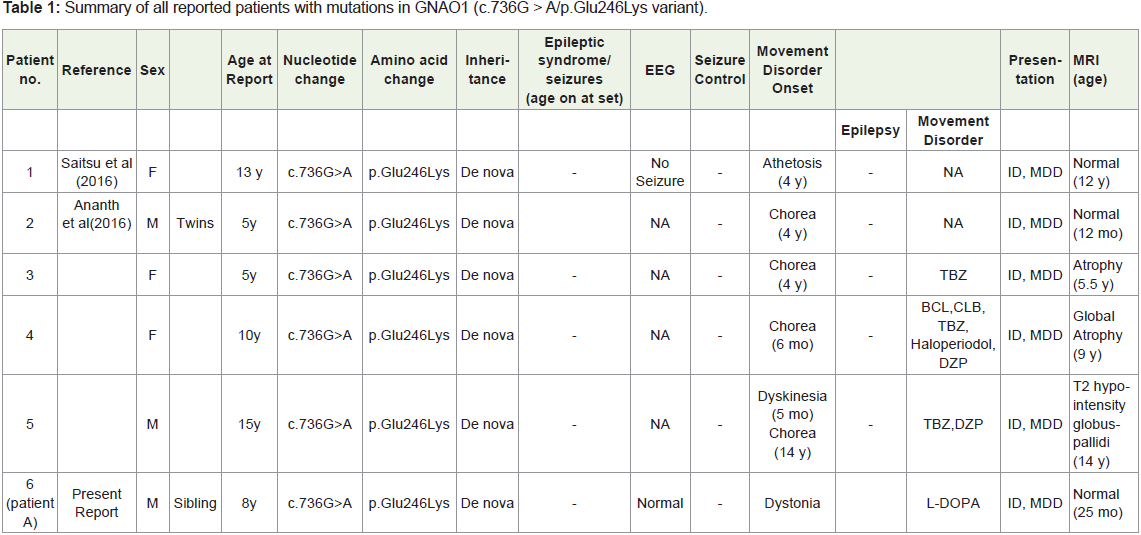
time of this report the de novo c.736G > A, p.Glu246Lys variant has
been reported in five other patients (four females, one male).
Discussion
GNAO1 encephalopathy is a rare neurodevelopmental disorder characterized by distinct movement presentations and early onset
epileptic encephalopathy. De novo GNAO1 mutations were originally
first reported in Ohtahara syndrome and EOEE in 4 children,
associated with abnormal movements in 2 children.
Since this initial description, 30 additional patients have
been reported with an emerging phenotype characterized by the
neurodevelopmental delay with an early onset of a hyperkinetic
movement disorder, inconsistently associated with epilepsy. The
occurrence of stereotypes (previously reported in 2 patients with
EOEE) and characteristic paroxysmal exacerbations, associated often
with clear triggers, may both be considered as 2 further important
discerning clinical features of GNAO1encephalopathy.
In this case report, it is evident that our patient predominately
presented with severe hyperkinetic movements with first episode of
clinical seizure activity at the age of 7 .The identified c.736G > A/p.
Glu246Lys variant has been reported in five other patients, so far none
of whom showed any epileptic seizures. Of note is that no differences
in phenotype were reported in affected twins of different sex carrying
this mutation, both showing chorea, but not seizures. Very recently
it was suggested that mutations affecting codons 209 or 246 of
GNAO1 specifically lead to a phenotype that involves involuntary
movement disorder and developmental delay but are characterized
by the absence of epilepsy. As the c.736G > A/p. Glu246Lys variant
would only affect transcript variant 1 of GNAO1 it appears possible
that a milder phenotype in regards to epilepsy results from a selective
relevance of transcript variants other than one that could result in
some functional GNAO1 in certain neuronal cell types.
There are some similarities as well as differences in the clinical
features of the present patient as well as previously reported cases.
Most of the previously reported patients had predominantly
hyperkinetic movements and only one had a focal seizures. The
commonly noted movement disorders were dystonia and chorea.
Athetoid movements were noted only in one patient. Our patient in
addition to dystonia had myoclonic jerks along with extensor spasms
which wasn’t reported in any of the previous patients with c.736G >
A/p.Glu246Lys variant. Though clinical seizures activity wasn’t the
predominant feature of previously reported cases with c.736G > A/p.
Glu246Lys variant our patient had one episode of focal tonic seizures
which is only the second case reported till date. (Table 1 summaries
all reported patients with mutations in GNAO1 with c.736G > A/p.
Glu246Lys variant).
With respect to the latest reports of patients showing a phenotype
with a predominant involuntary movement disorder, it appears
that the spectrum of symptoms is broader than initially suggested
so that mutations in GNAO1 should be considered in all patients
showing severe mental and motor retardation and either early
or severe epilepsy and/or some involuntary movement disorder.
Unfortunately, this is not always possible due to high costs of these
investigations, but on the other hand, the early etiological diagnosis
might reduce unnecessary investigations and might lead to a targeted
treatment reducing side effects from the trial-and-error treatment
approach. Taking into account the changing of the symptoms during
course of the disease, the long-term follow-up will eventually benefit
from knowing the basis of disease and its prognosis. Furthermore, it plays an important role in the process of informing the parents
about the cause of the disease and is of utmost importance in genetic
Counseling.
Acknowledgments
Authors would like to thank the family of the patients of the study
for extremely nice cooperation.
References
Citation
Satish S, Wilson R, Arunan S, Kalpana. A Rare Case of Movement Disorder with Developmental Delay [Clinical Phenotype of de novo Gnao1 mutation]: Case Report and Review of Literature. Indian J Neurol. 2022;3(1): 107.