Review Article
The Role of the Notch Pathway in Atherosclerosis
Cinzia Fortini1*, Cristiana Caliceti1, Giorgio Aquila1, Marco Bruno Morelli1, Rita Pavasini3 and Paola Rizzo1,2,4
Corresponding author: Dr. CinziaFortini, Department of Medical Sciences, University of Ferrara, Ferrara, Italy, Phone:+39 0532 242077,; E-mail: frtcnz1@unife.it
Citation: Fortini C, Caliceti C, Aquila G, Morelli MB, Pavasini R, et al. The Role of the Notch Pathway in Atherosclerosis. Indian J Cardio Biol Clin Sci. 2014;1(1): 103.
Copyright © 2015 Cinzia Fortini et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Indian Journal of Cardio Biology & Clinical Sciences | Volume: 2, Issue: 1
Submission: 31/10/2014; Accepted: 23/12/2014; Published: 24/12/2014
Abstract
Atherosclerosis is one of the primary causes of heart disease and stroke. In industrialized countries, it is the underlying cause of about 50% of all deaths. Atherosclerosis can be considered a form of chronic inflammation resulting from interaction between modified lipoproteins, monotype-derived macrophages, T cells and the cellular elements of the arterial wall. This inflammatory process can ultimately lead to the development of complex lesions or plaques that protrude into the arterial lumen. It is well established that the Notch signalling pathway regulates the functions of each cell type involved in the formation and in the evolution of the atherosclerotic plaques. In this review we will discuss the role of the Notch signalling in the modulation of macrophages, endothelial and vascular smooth muscle cells biological functions and how dysregulation of this pathway could potentially affect the onset and progression of atherosclerosis.
Introduction
Atherosclerosis is one of the leading cause of death worldwide. It is a multifactorial and multistep disease of the arterial wall that involves chronic inflammation not only during the initiation and evolution of lesions, but also during the development of acute thrombotic complications [1].
Atherosclerosis results from the complex interaction between vascular smooth muscle cells, the main cell type in the artery wall, and endothelial cells and macrophages [2]. The Notch pathway, a key regulator of cell fate, controls the biological functions of every cellular component of atherosclerotic plaque. Treatment of apolipoprotein E-deficient mice fed a high-fat diet with N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester(DAPT), an inhibitor of Notch signalling, reduces the number and size of plaques [3]. Similarly, Notch signalling inhibition by blocking Delta-like ligand 4 (Dll4), one of the ligands of the Notch receptors, attenuates the development of atherosclerosis, diminishes plaque calcification, improves insulin resistance and decreases fat accumulation in low-density lipoprotein (LDL)-receptor deficient mice [4]. These studies indicate that the Notch signalling pathway plays an important role in the onset and progression of atherosclerosis and therefore it could be a promising pharmacological target for the treatment of this pathology. This review will highlight the current knowledge on the role of the Notch signalling in the modulation of each cell type involved in atheroma formation and evolution.
Steps leading to atheromatous plaques formation
Atherosclerotic lesions are the result of highly specific cellular and molecular responses. The earliest changes that precede the formation of atherosclerosis take place in the endothelium. Changes in the endothelial cells permeability and in the composition of the extracellular matrix promote the entry and retention of cholesterolcontaining low density lipoprotein (LDL) particles in the artery wall [5] (Figure 1A). Monocytes from circulation penetrate into the endothelium, differentiate into macrophages, endocyte LDL particles and assume the morphology of foam cells that form the fatty streaks of the plaques of atheroma [2,6] (Figure 1B). Foam cells secrete matrix metallopeptidases, that promote vascular smooth muscle cells (VSMCs) migration from the media into the intima via the degradation of the extracellular matrix (ECM) (Figure 1C) [7]. In the intima, the VSMCs produce extracellular matrix molecules, including interstitial collagen and elastin, and form a fibrous cap that covers the plaque. This cap typically overlies a collection of macrophagederived foam cells, some of which die by apoptosis and release lipids that accumulate in the extracellular space. The inefficient clearance of dead cells, a process known as “efferocytosis”, promotes the accumulation of cellular debris and extracellular lipids, forming a lipid-rich pool called the necrotic core of the plaque (Figure 1D). The balance between cap and necrotic core influences the stability of the plaques. Stable plaques have a thick fibrous cap over a small fatty core and are rich in collagen. Conversely, unstable plaques have a thin fibrous cap over a large fatty core. Upon plaque rupture, the exposure of collagen, lipids, and smooth muscle cells leads to the activation of platelets and the coagulation cascade system and to the formation of a thrombus that may occlude the artery [8,9].

Figure 1: Atherosclerotic plaque formation.Alteration of endothelial cells permeability promotes lipids infiltration and accumulation (A). Monocytes from blood flow penetrate into the intima, differentiate inmacrophages and internalize lipids, forming foam cells (B). In response to pro-inflammatory cytokines and MMPs secretion,VSMCs proliferate and migrate (C),secrete extracellular matrix which form the fibrous cap of atheroma (D).
The Notch signalling pathway
The Notch signalling pathway has been highly conserved through evolution and it regulates cell-fate decisions through the embryonic development and during adult life [10]. In mammals there are four Notch receptors (Notch1-4) and five ligands (Dll1, 3, 4 and Jagged1 and 2). Both receptors and ligands are located on the cell surface and regulate communication between adjacent cells. Notch receptors are synthesized as single-chain precursors and cleaved in the Golgi apparatus into an extracellular and a transmembrane subunit by the proprotein processing enzyme furin. These two subunits are held together on cell membrane by non-covalent bonds. Activation of the Notch signalling pathway requires the binding of the Notch receptor to a ligand located on an adjacent cell. The endocytosis of the ligand, which follows the interaction with the receptor, provokes a mechanical force that alters the conformation of the Notch receptor ectodomain, making it susceptible to cleavage by A Disintegrin and Metalloprotease (ADAM). This proteolytic cut is followed by an intramembranous cleavage by γ-secretase, a multisubunit membrane protease which releases the active form of Notch, the Notch intracellular domain (NICD). NICD translocates to the nucleus, where it modulates the transcription of target genes in the receiving cells via recombinant signal binding protein 1 for Jκ (RPB-Jκ) transcription factor activation [10] (Figure 2). The most studied Notch target genes are the Hes (hairy and enhancer of split) and Hey (hairy related) gene families. Hes genes are crucial in neural and endocrine functions, Hey genes play a crucial role during the development of the cardiovascular system [11]. Other well-known Notch targets include p21Cip/Waf, cyclin D1, cyclin A and transcription factors of the NF-kB (nuclear transcription factor-kB) family. The set of directly and indirectly Notch-regulated genes and proteins is very large and new targets are continuously being discovered [12].

Figure 2: Notch pathwayPrecursor of Notch receptors undergoes the first cut in the Golgi by furin, which leads to the presentation of the receptor on the membrane in a functionalheterodimeric form. Upon interaction with a ligand present on the surface of adjacent cells, ADAM performs the second cut. The third cut, which promotes therelease of the intracellular domain of the receptor, is made by the γ-secretase complex. The resulting fragment translocates into the nucleus where it promotesthe transcription of Notch target specific genes such as Hes1, Hey1 and 2.
Notch activity is tightly regulated by post-translational modifications such as phosphorylation, glycosylation, rapid ubiquitination-mediated degradation [12], and bycross-talks with inflammatory cytokines (10) as well as with other key pathways such as NF-kB, estrogen receptor α, ErBB2 [13] and vascular endothelium growth factor receptors (VEGFRs) [14]. As a result of this complexnetwork of interactions, the effects of Notch activation (or inhibition) are strongly dependent on the cellular context and by timing,duration, and levels of activation (or inhibition).
Role of Notch signalling in cellular componentsinvolved in atherogenesis
Role of Notch in endothelial cells
The role of Notch pathway in the development of the vascular system is well established [15]. Mouse embryos carrying mutations of Notch1 or both Notch1 and 4 showed severe lethal vascular defects [15]. Molecular data indicate that during development, Notch acts mainly by determining arterial-venous specification: specifically, active Notch signalling will suppress venous cell fate and promotearterial determination [15,16].
Notch receptors 1, 2, 3 and 4, Dll1, 4 and Jagged1, 2 ligands are all expressed in the adult vasculature [17], in which Dll4/Notch1- mediated signalling modulates VEGF-driven angiogenesis by regulating the number of sprouts (new branches) on endothelial cells. According to a well-established model, the interplay between Dll4/Notch1/VEGFR2 determines the balance between the number of tip cells (able to direct the blood vessel sprout and characterized by low Notch1 and high VEGFR2) and stalk cells (proliferating cells forming the vascular lumen, characterized by high Notch and low VEGFR2) [18]. Notch ligand Jagged1 is a potent pro-angiogenic regulator in mice that antagonizes Dll4-Notch1 signalling in cells expressing Fringe family glycosyl transferases [19]. Recent published workhas shown that Notch-dependent activation of VEGFR3 promotes angiogenesis even in the absence of VEGF-VEGFR2 signalling [20].
Endothelial cells dysfunctions are the first step towards the onset of atheromatous plaques. Endothelial cells dysfunction is a broad term that implies increased expression of adhesion proteins for blood leukocytes, increased permeability, reduced expression of endothelial nitric oxide synthase (eNOS), increased apoptosis and reduced ability to proliferate in the presence of an endothelium damage [21]. Notch plays an important role in protecting endothelial cells from apoptosis. In vitro and in vivo treatment with inflammatory cytokines TNF-α and IL-1β induces the dysregulation of Notch signalling (downregulation of Notch4 and increased expression of Notch2) which is associated to activation of the NF-kB pathway and to increased levels of ICAM-1 (intercellular adhesion molecule-1), VCAM-1 (vascular cell adhesion molecule-1) and of apoptosis [22,23]. A prominent role for Notch4 has been shown in cardiac allograft vessels, in which impaired Notch4 expression in graft endothelial cells is a key event associated with transplant atherosclerosis by triggering EC activation and apoptosis [24]. Recent data provide evidence that NF-kB is a negative regulator of eNOS expression via upregulation of miR-155 [25] and Notch signalling represses miR-155 expression by promoting the binding of RBP-Jk to the miR-155 promoter [26]. Loss of Notch/ RBP-Jk signalling upregulates miR-155 in bone marrow endothelial cells, leading to miR-155-mediated targeting of NF-kB inhibitor kBras1, NF-kB activation, and increased pro-inflammatory cytokines production [26] (Figure 3A).
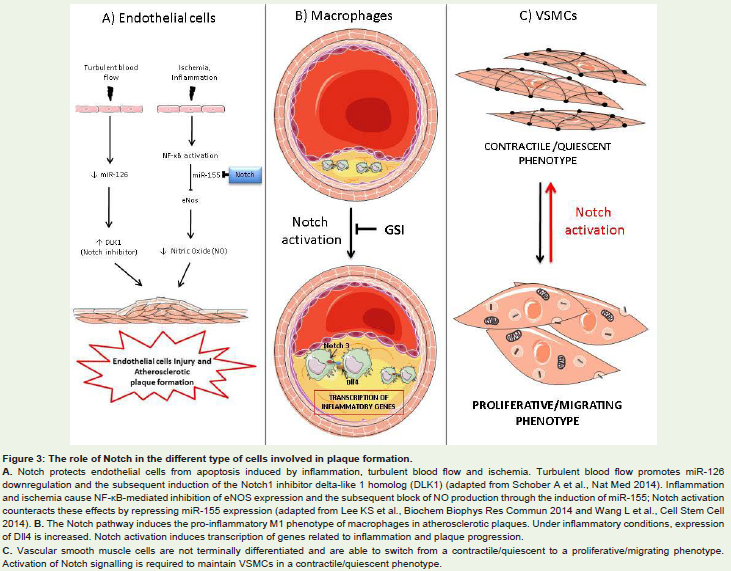
Figure 3: The role of Notch in the different type of cells involved in plaque formation.A. Notch protects endothelial cells from apoptosis induced by inflammation, turbulent blood flow and ischemia. Turbulent blood flow promotes miR-126downregulation and the subsequent induction of the Notch1 inhibitor delta-like 1 homolog (DLK1) (adapted from Schober A et al., Nat Med 2014). Inflammationand ischemia cause NF-κB-mediated inhibition of eNOS expression and the subsequent block of NO production through the induction of miR-155; Notch activationcounteracts these effects by repressing miR-155 expression (adapted from Lee KS et al., Biochem Biophys Res Commun 2014 and Wang L et al., Cell Stem Cell2014). B. The Notch pathway induces the pro-inflammatory M1 phenotype of macrophages in atherosclerotic plaques. Under inflammatory conditions, expressionof Dll4 is increased. Notch activation induces transcription of genes related to inflammation and plaque progression.C. Vascular smooth muscle cells are not terminally differentiated and are able to switch from a contractile/quiescent to a proliferative/migrating phenotype.Activation of Notch signalling is required to maintain VSMCs in a contractile/quiescent phenotype.
Conditions of disturbed blood flow existing at bifurcations and at bends of the arteries (i.e. the aortic arch and carotids bifurcations) cause endothelial dysfunctions by inducing a pro-inflammatory and pro-apoptotic gene expression profile and make these regions prone to atheromas formations (athero-prone). Specifically, in these areas NF-kB is primed for activation [21] and there is expression of marker of endothelial cells activation and a higher number of apoptotic cells in comparison with athero-protected areas [27]. The Notch pathway is one of the many pathways regulated by the hemodynamic forces exerted by the blood flow on the endothelium. Exposure of endothelial cells of the microvasculature to laminar blood flow promotes the expression of Notch1 which, in turn, protects cells from apoptosis through the induction of anti-apoptotic protein Bcl-2 [28]. The link between hemodynamic forces and Notch signalling has been confirmed by in vivo studies in zebrafish embrios [29]. Futhermore, Schober et al. have recently shown that downregulation of endothelial miR-126-5p by disturbed blood flow in proatherogenic region of aortic arch, abrogates endothelial cells proliferation in response to hyperlipidemic stress through upregulation of the Notch1 inhibitor delta-like 1 homolog (DLK1) [30] (Figure 3A).
It is thought that endothelial progenitor cells (EPCs) contribute to re-endothelization and play a role in the neo-vascularization of ischemic tissues and in tissue repair. Consistently, it has been reported that the number of circulating EPCs increases in patients with cardiovascular disease [31] and it is reduced in diabetic patients with diabetic foot syndrome [32]. Jagged1-mediated Notch signalling activation from the bone marrow microenvironment is critical for EPCs mobilization and angiogenesis [33]. One of the mechanisms by which Notch-RBP-Jk signalling regulates the mobilization and function of EPCs is by modulating the expression of chemokine receptor 4 (CXCR4), the receptor for stromal derived factor 1 (SDF- 1) involved in EPCs chemotaxis [34].
Role of Notch in macrophages
Activated macrophages participate in every stage of atherosclerotic lesion progression, from fatty streak formation to the onset of acute thrombotic complications. Matrix-degrading enzymes and prothrombotic molecules secreted by activated macrophages promote plaque disruption and subsequent thrombosis [9,35].
Notch3 mRNA selectively increases during the differentiation of human monocytes to macrophages [36] and resting macrophages express Notch 1, 2, and 4, as well as the Notch ligands Jagged 1 and 2 as observed both in monocyte cell lines and in mouse peritoneal macrophages [37].
Notch3 and Dll4 are expressed in macrophages of humancarotid atherosclerotic lesions (36) and several evidences indicate that in macrophages the Notch pathway contributes to the induction of a pro-inflammatory status. Treatment of macrophages with pro-inflammatory stimuli, such as TNF-α, IL-1β or LPS (Lipopolysaccharides), induces the activation of Notch1, Jagged1 [37] and Dll4 [36]. The activation of Notch signalling leads to the transcription of inflammatory genes, such as inducible nitric oxide synthase (iNOS), pentraxin 3, DNA-binding protein inhibitor ID-1(Id1), and promotes the activation of Akt and of extracellular signal-regulated kinases 1 and 2 (ERK 1 and 2) pathways. Moreover, the increase of Dll4 expression correlates with a reduction of IκBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor α) which leads to activation of the NF-κB pathway [36](Figure 3B).
In response to various stimuli, macrophages switch from the M1 (classical, pro-inflammatory) to the M2 (alternative, antiinflammatory) phenotype. M1 macrophages are induced by IL- 1β and TNF-α, while M2 are stimulated by IL-4 and IL-10 [38]. Notch signalling regulates macrophage polarization by inducing the transcription factor Interferon regulatory factor 8 (IRF8) involved in induction of M1 macrophage–associated genes [39]. In particular, Dll4 seems to play a pivotal role in shifting macrophages toward a pro-inflammatory phenotype. In vitro studies have shown that the blockade of Dll4 expression using specific siRNA reduces the expression of typical pro-inflammatory M1 mediators (e.g., iNOS,TNF-α and IL-1β) and increases the expression of mannose receptor 1 (MRC1) and IL-10, markers of M2 macrophage polarization [4].
In vivo studies have shown that Notch activation favours the progression of atherosclerosis. Quantitative RT-PCR analyses conducted on thoracic aortas RNA isolated from apolipoprotein E-deficient (ApoE-/-) mice fed an atherogenic diet revealed higher expression of Notch1, Notch3, Jagged1 and Hes1 in comparison with wild type mice [3]. In order to test if Notch inhibition could limit the progression of atherosclerosis, in the same study, ApoE-/-) mice were treated with the γ-secretase inhibitor (GSI) LY411,575. Histological analysis of atherosclerotic plaque isolated from treated mice showed that the area of the lesion was highly reduced compared with untreated mice [3]. Furthermore, immunostaining of macrophages isolated from peritoneal cavity of mice treated with LY411,575, showed a reduction in the expression levels of ICAM-1, of Hes1 and in the number of atherosclerotic plaque-localized macrophages in comparison with control mice [3]. These data are consistent with previous work showing that Notch1 signalling upregulates the expression of ICAM-1 in M1 macrophages [37]. Furthermore, increased levels of Dll4 were found in the aorta and in adipose tissue of LDL-receptor-deficient mice fed a high fat diet, compared with mice fed a standard diet [4]. Treatment of LDL-receptor-deficient mice with an antibody against Dll4 reduced not only aortic expression of the Notch target gene Hey2 but also the size of atherosclerotic lesions, the expression of monocyte chemoattractant protein-1 (MCP-1) as well as the accumulation of macrophages in aortic lesions.
In conclusion, the Notch signalling pathway may contribute to the progression of atherosclerosis by promoting a M1-macrophage phenotype and therefore by amplifying the inflammatory response within the atheromatous plaques.
Role of Notch in vascular smooth muscle cells
In addition to the macrophages, VSMCs are important components of atherosclerotic plaques. In particular, the balance between proliferation and apoptosis of VSMCs has an impact on the intimal thickeness and it may also affect atherosclerotic plaque stability [1,7]. Moreover, VSMCs proliferation and migration is one of the major causes of restenosis in a fraction of patients that undergoes angioplasty or stent application [40]. In the mature blood vessel, VSMCs exhibit differentiated phenotype characteristics, such as the expression of contractile markers specific to smooth muscle, thea ssembly of actin cytoskeleton into parallel and the elongatedstress fibres. However, VSMCs retain remarkable plasticity and can undergo rather profound phenotypic modifications in response to changes in local environmental cues [41]. VSMCs switch from a contractile/quiescent to a secretory/inflammatory/migratory state in response to pro-inflammatory cytokines, such as TNF-α and IL- 1β and migrate from the media to the intima, where they synthesize extracellular matrix macromolecules such as collagen, elastin and proteoglycans.
Notch is one of major signalling pathways involved in controlling VSMCs survival, phenotypic modulation and migration. Adult rat VSMCs express Notch1 and Notch3 and the target genes Hes1 and Hes5 [42]. In rat VSMCs, inhibition of Notch using RPMS-1 (an Epstein-Barr virus encoded gene product that inhibits CBF-1/RBP-Jk, the trascription factor bound by Notch) or brefeldin A (an inhibitor of the cellular transport from the endoplasmic reticulum to the Golgi apparatus), induces a significant increase in apoptosis whereas stable expression of the active form of Notch1 and 3 promotes cells survival [42].
The acquisition of a secretory phenotype in VSMCs induced by proinflammatory stimuli is mediated by the increase of adenylyl cyclase 8 (AC8) expression. Adenylyl cyclases, along with phosphodiesterases, regulates conversion of ATP into cyclic adenosine monophosphate (cAMP). During the VSMCs trans-differentiation process, increased expression of AC8 protein occurs, modulating cAMP signalling and controlling the ability of VSMCs to acquire properties specific to the secretory state [43]. In rat VSMCs treated with IL-1β, an increase in the expression levels of AC8 was observed, which was counteracted by Notch3 overexpression. Conversely, treatment with Notch inhibitor DAPT enhanced the effect of IL-1β on AC8 expression [44]. Studies in rat carotid arteries after balloon injury, have confirmed that Notch3 negatively regulates AC8 expression [44].
Experiments conducted in rat aortic smooth muscle cells have shown that pro-inflammatory cytokines promote the transition of VSMCs from a contractile to a secretory phenotype by NF-kB– mediated inhibition of the Notch pathway [45]. Specifically, Clement et al. have shown that Dll1-mediated activation of Notch receptors or the overexpression of the active form of Notch3 decreases IL- 1β mRNA expression levels and the secretion of the inflammatory marker phospholipase A2 (PLA2). Vice versa, treatment of VSMCs with IL-1β reduces mRNA expression levels of Jagged1 and of the Hey family target genes. Blocking the NF-kB pathway with a mutated form of IkBα restores Notch pathway components expression [45](Figure 3C).
VSMCs migration to the injury site requires the secretionof matrix metallopeptidases (MMPs) for the degradation of the extracellular matrix. In vitro studies have shown that plaque resident VSMCs and macrophages release MMP1, 3 and 9 in response to growth factors and inflammatory cytokines [46]. Treatment of rat aortic VSMCs with docosahexaenoic acid (DHA, a polyunsaturated fatty acid with anti-inflammatory effects), prevents the proliferative effect induced by pro-inflammatory stimuli and reduces the level of MMP2 and 9. Noteworthy, in these cells over-expression of the active form of Notch1 and 3 counteracted IL-1β-mediated effect on expression of MMP2 and 9. Conversely, Notch inhibition increased over-expression of MMP 2 and 9 transcripts in rat aortic VSMCs induced by IL-1β [47].
Taken together these data indicate that the Notch pathway, by counteracting the effects of inflammatory cytokines on VSMCs, promotes atherosclerotic plaques stability.
Conclusions
The Notch pathway plays a pivotal role in atherosclerotic plaques formation and progression, acting on every cellular type involved in this process. Overall, Notch signalling in macrophages seems to favor pro-inflammatory responses whereas in endothelial and vascular smooth muscle cells Notch antagonizes the effects of inflammation. Given the complex role played by Notch in the context of atherosclerosis, more studies are needed to understand whether the manipulation of this pathway could offer a new therapeutic approach to interfere with atherosclerosis progression.
References
- Libby P, Ridker PM, Hansson GK 2011 Progress and challenges in translating the biology of atherosclerosis. Nature 473: 317-325.
- Hansson GK, Hermansson A 2011 The immune system in atherosclerosis. Nat Immunol 12: 204-212.
- Aoyama T, Takeshita K, Kikuchi R, Yamamoto K, Cheng XW, et al. 2009 gamma-Secretase inhibitor reduces diet-induced atherosclerosis in apolipoprotein E-deficient mice. Biochem Biophys Res Commun 383: 216-221.
- Fukuda D, Aikawa E, Swirski FK, Novobrantseva TI, Kotelianski V, et al. 2012 Notch ligand delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc Natl Acad Sci U S A 109: E1868-E1877.
- Tabas I, Williams KJ, Boren J 2007 Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 116: 1832-1844.
- Hansson GK 2001 Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol 21: 1876-1890.
- Newby AC 2006 Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc Res 69614-624.
- Tabas I 2010 Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 10: 36-46.
- Aikawa M, Libby P (2004) The vulnerable atherosclerotic plaque: pathogenesis and therapeutic approach. Cardiovasc Pathol 13: 125-138.
- 10. Rizzo P, Miele L, Ferrari R (2013) The Notch pathway: a crossroad between the life and death of the endothelium. Eur Heart J 34: 2504-2509.
- Wiese C, Heisig J, Gessler M (2010) Hey bHLH factors in cardiovascular development. Pediatr Cardiol 31: 363-370.
- Espinoza I, Miele L (2013) Notch inhibitors for cancer treatment. Pharmacol Ther 139: 95-110.
- Rizzo P, Osipo C, Pannuti A, Golde T, Osborne B, Miele L (2009) Targeting Notch signaling cross-talk with estrogen receptor and ErbB-2 in breast cancer. Adv Enzyme Regul 49: 134-141.
- Gu JW, Rizzo P, Pannuti A, Golde T, Osborne B, et al. (2012) Notch signals in the endothelium and cancer "stem-like" cells: opportunities for cancer therapy. Vasc Cell 4: 7.
- Kume T (2010) Specification of arterial, venous, and lymphatic endothelial cells during embryonic development. Histol Histopathol 25: 637-646.
- Sorensen I, Adams RH, Gossler A (2009) DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood 113: 5680-5688.
- Lindner V, Booth C, Prudovsky I, Small D, Maciag T, et al. (2001) Members of the Jagged/Notch gene families are expressed in injured arteries and regulate cell phenotype via alterations in cell matrix and cell-cell interaction. Am J Pathol 159: 875-883.
- Jakobsson L, Bentley K, Gerhardt H (2009) VEGFRs and Notch: a dynamic collaboration in vascular patterning. Biochem Soc Trans. 37: 1233-1236.
- Benedito R, Roca C, Sorensen I, Adams S, Gossler A, et al. (2009) The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137: 1124-1135.
- Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, et al. (2012) Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 484: 110-114.
- Iiyama K, Hajra L, Iiyama M, Li H, DiChiara M, et al. (1999) Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ Res 85: 199-207.
- Quillard T, Devalliere J, Chatelais M, Coulon F, Seveno C, et al. (2009) Notch2 signaling sensitizes endothelial cells to apoptosis by negatively regulating the key protective molecule survivin. PLoS One 4: e8244.
- Quillard T, Devalliere J, Coupel S, Charreau B (2010) Inflammation dysregulates Notch signaling in endothelial cells: implication of Notch2 and Notch4 to endothelial dysfunction. Biochem Pharmacol 80: 2032-2041.
- Quillard T, Coupel S, Coulon F, Fitau J, et al. (2008) Impaired Notch4 activity elicits endothelial cell activation and apoptosis: implication for transplant arteriosclerosis. Arterioscler Thromb Vasc Biol 28: 2258-2265.
- Lee KS, Kim J, Kwak SN, Lee KS, Lee DK, et al. (2014) Functional role of NF-kappaB in expression of human endothelial nitric oxide synthase. Biochem Biophys Res Commun 448: 101-117.
- Wang L, Zhang H, Rodriguez S, Cao L, Parish J, et al. (2014) Notch-dependent repression of miR-155 in the bone marrow niche regulates hematopoiesis in an NF-kappaB-dependent manner. Cell Stem Cell 15: 51-65.
- Nigro P, Abe J, Berk BC (2011) Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid Redox Signal 15: 1405-1414.
- Walshe TE, Connell P, Cryan L, Ferguson G, Gardiner T, et al. (2011) Microvascular retinal endothelial and pericyte cell apoptosis in vitro: role of hedgehog and Notch signaling. Invest Ophthalmol Vis Sci 52: 4472-4483.
- Watson O, Novodvorsky P, Gray C, Rothman AM, Lawrie A, et al. (2013) Blood flow suppresses vascular Notch signalling via dll4 and is required for angiogenesis in response to hypoxic signalling. Cardiovasc Res 100: 252-261.
- Schober A, Nazari-Jahantigh M, Wei Y, Bidzhekov K, Gremse F, et al. (2014) MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1.Nat Med 20: 368-476.
- Campioni D, Zauli G, Gambetti S, Campo G, Cuneo A, et al. (2013) In vitro characterization of circulating endothelial progenitor cells isolated from patients with acute coronary syndrome. PLoS One 8:e56377.
- Drela E, Stankowska K, Kulwas A, Rosc D (2012) Endothelial progenitor cells in diabetic foot syndrome. Adv Clin Exp Med 21: 249-254.
- Kwon SM, Eguchi M, Wada M, Iwami Y, Hozumi K, et al. (2008) Specific Jagged-1 signal from bone marrow microenvironment is required for endothelial progenitor cell development for neovascularization. Circulation 118: 157-165.
- Wang L, Wang YC, Hu XB, Zhang BF, Dou GR, et al. (2009) Notch-RBP-J signaling regulates the mobilization and function of endothelial progenitor cells by dynamic modulation of CXCR4 expression in mice. PLoS One 4:e7572.
- Shah PK (2003) Mechanisms of plaque vulnerability and rupture. J Am Coll Cardiol 41: 15S-22S.
- Fung E, Tang SM, Canner JP, Morishige K, Arboleda-Velasquez JF, et al. (2007) Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation 115: 2948-2956.
- Monsalve E, Perez MA, Rubio A, Ruiz-Hidalgo MJ, Baladron V, et al. (2006) Notch-1 up-regulation and signaling following macrophage activation modulates gene expression patterns known to affect antigen-presenting capacity and cytotoxic activity. J Immunol 176: 5362-5373.
- Martinez FO, Gordon S, Locati M, Mantovani A (2006) Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 177: 7303-7311.
- Xu H, Zhu J, Smith S, Foldi J, Zhao B, et al. (2012) Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat Immunol 13: 642-650.
- Hao H, Gabbiani G, Bochaton-Piallat ML (2003) Arterial smooth muscle cell heterogeneity: implications for atherosclerosis and restenosis development. Arterioscler Thromb Vasc Biol 23: 1510-1520..
- Clarke M, Bennett M (2006) The emerging role of vascular smooth muscle cell apoptosis in atherosclerosis and plaque stability. Am J Nephrol 26: 531-535.
- Sweeney C, Morrow D, Birney YA, Coyle S, Hennessy C, et al. (2004) Notch 1 and 3 receptor signaling modulates vascular smooth muscle cell growth, apoptosis, and migration via a CBF-1/RBP-Jk dependent pathway. FASEB J 18: 1421-1423.
- Gueguen M, Keuylian Z, Mateo V, Mougenot N, Lompre AM, et al. (2010) Implication of adenylyl cyclase 8 in pathological smooth muscle cell migration occurring in rat and human vascular remodelling. J Pathol 221: 331-342.
- Keuylian Z, de Baaij JH, Gueguen M, Glorian M, Rouxel C, et al. (2012) The Notch pathway attenuates interleukin 1beta IL1beta-mediated induction of adenylyl cyclase 8 AC8 expression during vascular smooth muscle cell VSMC trans-differentiation. J Biol Chem 287: 24978-24989.
- Clement N, Gueguen M, Glorian M, Blaise R, Andreani M, et al. (2007) Notch3 and IL-1beta exert opposing effects on a vascular smooth muscle cell inflammatory pathway in which NF-kappaB drives crosstalk. J Cell Sci 120: 3352-3361.
- Bond M, Chase AJ, Baker AH, Newby AC (2001) Inhibition of transcription factor NF-kappaB reduces matrix metalloproteinase-1, -3 and -9 production by vascular smooth muscle cells. Cardiovasc Res 50: 556-565.
- Delbosc S, Glorian M, Le Port AS, Bereziat G, Andreani M, et al. (2008) The benefit of docosahexanoic acid on the migration of vascular smooth muscle cells is partially dependent on Notch regulation of MMP-2/-9. Am J Pathol 172: 1430-1440.