Case Report
Allgrove Syndrome without Achalasia: A Rare Case Report and Brief Review of Literature
Vasishta Reddy S1, VB Kasyapa Jannabhatla2*, Sunanda Tirupathe2 and Bhimeswara Rao P3
1Department of Radiodiagnosis, Katuri Medical College & Hospital, Guntur, Andhra Pradesh, India
2Department of Endocrinology, Narayana Medical College & Hospital, Nellore, Andhra Pradesh, India
3Department of Radiodiagnosis, Katuri Medical College & Hospital, Guntur, Andhra Pradesh, India
2Department of Endocrinology, Narayana Medical College & Hospital, Nellore, Andhra Pradesh, India
3Department of Radiodiagnosis, Katuri Medical College & Hospital, Guntur, Andhra Pradesh, India
*Corresponding author: V B Kasyapa Jannabhatla, Department of Endocrinology, Narayana Medical College & Hospital, Nellore, Andhra Pradesh, India, Email ID: drvbkasyapa@gmail.com; Mobile no: 8686866568
Copyright: © 2023 Vasishta Reddy S, et al. This is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Article Information: Submission: 15/03/2023; Accepted: 08/06/2023; Published: 12/06/2023
Abstract
Since the first description by Allgrove at al. Triple A syndrome (Allgrove syndrome) was reported rarely with an estimated prevalence of 1 in 1,000,000 individuals. It is a progressive degenerative disease comprising absent tear production (alacrima), achalasia of cardia, and adrenal insufficiency, caused by mutations in AAAS gene on chromosome 12q13. ALADIN protein is a part of nuclear pore complex with significant functional implications in many
tissues. We present a 9-year 9-month old girl presenting with hypoglycemic seizures and hyperpigmentation, later found to have adrenal insufficiency. Upon investigations alacrima, and partial optic atrophy were found and Allgrove syndrome was suspected. Interestingly our case was not associated with the
common presentation of achalasia, but has primary hypothyroidism which is rarely reported in association. Genetic report revealed a homozygous mutation in exon 6 of AAAS gene causing truncating protein production confirming the diagnosis. Allgrove syndrome is often diagnosed late, emphasizing the need for a high index of suspicion for alacrima and glucocorticoid insufficiency symptoms. Thorough biochemical and radiological investigations are recommended in suspected cases. Genetic testing can aid in diagnosis and provide valuable information for genetic counseling and follow-up planning. The syndrome presents with a wide range of symptoms, including achalasia, adrenal insufficiency, alacrima, and various neurological manifestations. This case highlights the importance of recognizing and managing the multisystemic features of the syndrome.
Keywords: Allgrove syndrome; AAA Syndrome; Alacrima; Achalasia; 4A Syndrome; Aladin Protein
Introduction
Glucocorticoid deficiency associated with achalasia of the cardia
and absent tear production was described by Allgrove et al. It was
first described in two pairs of siblings in 1978 [1].The estimated
prevalence is 1 per 1,000,000 individual [2]. Lanes et al. suggested that
a progressive degenerative process may be responsible for the three
components of the disease and coined the term ‘triple-A syndrome’
(AAAS) [3]. Gazarian et al. referred this syndrome as ‘4A syndrome,’
associating various autonomic and neurologic disturbances (ND) as
components [4]. In 1996, Weber et al.[5] localized the disease gene on chromosome 12q13. The gene produces alacrima-achalasia-adrenal
insufficiency neurologic disorder (ALADIN) protein, which belongs
to the WD-repeat family of proteins. Clark and Weber et al. pointed
out that the AAAS has variable phenotypic expression. The triple-A
gene, designated AAAS, was cloned by Tullio-Pelet et al [6]. And
Hands chug et al [7] in 2000.
ALADIN protein belongs to the WD-repeat family of regulatory
proteins, with functions ranging from transmembrane signaling
and transcription, to cell division and intracellular trafficking [7,8].
Missense, nonsense and splicing mutations in AAAS gene cause the
protein to mislocalize to the cytoplasm[9]. Cells from subjects with
Allgrove syndrome do not show morphologic abnormalities in the
nuclei, nuclear envelope or nuclear pore complexes, suggesting that
mutations in AAAS gene result in functional, rather than structural, abnormalities in the nuclear pore complex [10]. Frame shift, stop
codon and functionally significant mutations lead to a more severe
phenotype, probably occurring by a loss of function effect on the
protein [11-14]. Mutations of the AAAS gene cannot be
identified in all clinically diagnosed AAAS patients [7,10,13,15].
A progressive neurological syndrome with central, peripheral and
autonomic nervous system impairment, often associated with mild
intellectual disability, has been described [11-14].
Case
A 9-year 9-month old female child was referred from pediatric
emergency for hypoglycemic seizures. The patient was apparently
normal 5 years back. The parents observed darkening of the body with
frequent fever, vomiting, abdominal pain, and seizures for a year.
She was diagnosed with hypothyroidism and a seizure disorder
and was prescribed levothyroxine and Valproate. Valproate was
discontinued after the diagnosis of Addisons disease. Hydrocortisone
was added, and later she discontinued the drugs. Now she has
presented with similar complaints for the past 5 days.
Careful history revealed that she had “always cried without tears”,
generalised blackening of skin, recurrent vomiting and seizures. She
did not have the regurgitation of undigested food. Her birth history
was normal without delayed milestones or speech. She was born out
of non-consanguineous marriage and no similar complaints were
seen in the family, including her younger sibling, who is 8 years old.
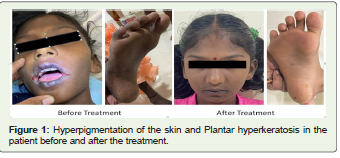
On examination she was malnourished, irritable, and had mild
intellectual disability, generalised hyper pigmentation, hyperkeratosis
of the palms and soles, pre-pubertal secondary sexual characters,
normal blood pressure, tachycardia, and mildly exaggerated tendon
jerks. Her height and weight were between the 10-25th centile for
her age. No skeletal abnormalities, ambiguous genetalia, cognitive
impairment, or other autonomic disturbances were observed.
Investigations showed Mild to moderate intellectual disability,
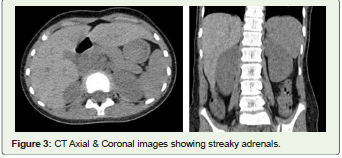
low serum cortisol, high ACTH, <1 mm Schirmer test score, bilateral
partial optic atrophy, and bilateral thin and streaky adrenals on CT
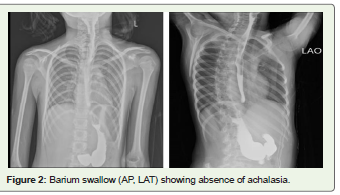
abdomen. Barium swallow did not show achalasia.
In view of alacrima, adrenal insufficiency, and optic atrophy
the clinical diagnosis of Allgrove syndrome was considered and
hydrocortisone was restarted. Patient has shown significant
improvement. Levothyroxine was continued. Methylcellulose eye
drops were added. Clinical exome sequencing was suggested.

Genetic report has shown a pathogenic variant of Allgrove
syndrome which is a homozygous nonsense variant in exon 6 of
AAAS gene on chromosome 12 that results in a stop codon and
premature truncation of the protein at codon 230 (p.Ard230Ter).
Genetic counseling was given to parents and regular follow up was
advised. We have obtained written informed consent from the patient
for reporting this rare case.
Discussion
The age of presentation falls within the first two decades of life
(1.3 - 13.8 years) with median age being 5 years [2]. While age at
presentation and diagnosis is early in familial cases, the etiological
diagnosis is usually delayed until 11.4 years of age. The delay is
attributed to the lack of detailed clinical characterization of the
disease [16,17]. Our case was presented at the age of 6 years to an
outside hospital, and the genetic diagnosis was established at the age
of 10 years.
The majority of patients present with the symptoms of achalasia
and adrenal insufficiency, while other presenting complaints included
seizures, poor growth, hyper pigmentation of the skin, developmental
delay, and autonomic disturbances[18,19]. Other associated symptoms
included alacrima, Palmoplantar hyperkeratosis, optic atrophy, ataxia,
intellectual disability, dysmorphic facies, microcephaly, sensory
neural deafness, hypotonia, vitiligo, and neuromuscular dystrophy
[20,21]. In our case the presenting complaint was a hypoglycemic
seizure which was reported to be present in 10-36% of cases [17],
even though the symptom of alacrima has been present since birth.
Palmoplantar hyperkeratosis, mild intellectual disability, and hyper
pigmentation of the skin are also present in our case.
Alacrima is the earliest and most consistent finding which is
often overlooked by parents with prevalence reaching >90% in the
affected patients[18,21]. It is present since birth or early infancy [16].Parasympathetic dysfunction may be the cause for loss of tear
production and may lead to punctiform corneal destruction [21]. It is
diagnosed by Schirmer’s test. Orbital MRI/CT is indicated if patient
is not cooperative. T1- and T2-weighted MR images are similar to
those of the extra ocular muscles and cerebral gray matter for lacrimal
glands in lacrimal fossa [22]. They are often atrophic or absent on
orbital CT images. Biopsy reveals neuronal degeneration and
depletion of secretory granules in the acinar cells [23]. Administration
of artificial tears and lubricants to relieve dryness are indicated.
Alacrima may lead to corneal ulceration and keratopathy. Visual
acuity assessment, ocular surface study, tonometry, and fundus
examination are recommended at least yearly [21]. In our patient we
diagnosed alacrima through Schirmer’s test and prescribed lubricant
eye drops. Partial optic atrophy present in our patient did not affect
the vision. Other ophthalmic manifestations reported were kerato
conjunctivitis sicca, papillary abnormalities, including sluggish
pupils, hypersensitivity to dilute miotics, optic atrophy, and steroid
induced lamellar cataracts in posterior subcapsule [17].
Adrenal insufficiency (AI) is seen in 80-100% of the patients.
While 85% of the cases show Glucocorticoids deficiency, only 15%
of cases show mineralcorticoid deficiency [23]. Glucocorticoid
deficiency can be borderline with normal ACTH-stimulation
test [13]. Dysfunction of the ALADIN protein has been shown to
down-regulate genes encoding cytochrome P450 hydroxylases, and
oxidoreductases, leading to reduced synthesis of precursors of steroid
hormones [24]. Low cortisol, and DHEAS levels are seen in majority
of the patients [17]. Similar to the literature, the adrenal insufficiency
in our case manifested as episodic hypoglycemia-induced seizures and
progressive hyper pigmentation. A 6 monthly follow up with stress
dosing of steroid therapy is recommended in these patients. Normal
electrolytes in our case excludes mineralocorticoid deficiency.
Achalasia cardia is seen in 75-100% of the cases and usually a first
symptom for seeking medical attention [19]. It is rare in children,
but in AAA syndrome, it presents in first decade of life. Achalasia
typically presents with vomiting, swallowing difficulties, weight loss,
and chronic cough, but it can also present as tooth decay[17,25].
Neuronal nitric oxide synthase deficiency due to oxidative damage
to ganglionic cells and nerve fibers in lower esophagus explains the
poor relaxation of the lower esophageal sphincter [26]. HRCT chest
may show tracheobronchial compression, and lung parenchymal
lesions[27]. Manometry is the gold standard diagnostic test.
Pneumatic lower esophageal dilatations, medical treatment with
calcium channel blockers, botulinum toxin, nitrates, and heller’s
cardiomyotomy are the main stay of treatments [21]. In contrast to
many studies which has shown early achalasia with truncated protein
mutations, the absence of symptoms and negative barium swallow
study excluded the achalasia in our patient.
Around 30% of AAAS patients suffer from autonomic
impairment along with other neurological manifestations [19].
Postural hypotension, abnormal sweating, tachycardia, and
hyperreflexia are the most common neurological abnormalities
[17,18]. Other autonomic dysfunction symptoms are arrhythmias,
anisocoria, abnormal papillary responses, and impotence [21].
Hypernasal speech, ataxia, sensory impairment, bulbospinal
amyotrophy, parkinsonian features, cranial nerve manifestations,
Chiari 1 malformation, and optic atrophy are the other important
neurological symptoms [2,28]. MRI/CT of head and spine are usually
normal [13]. They develop slowly and presents at a later stage of life
[17]. Glucocorticoid supplementation does not affect the progression
of the symptoms. Muscle biopsies show neurogenic degeneration,
non-specific myopathy, or mixed pathologies [21]. Our patient has
already shown partial optic atrophy, hyperreflexia, and tachycardia.
Due to the uncooperative patient, MRI brain was not performed.
However outside CT brain was normal.
Although thyroid dysfunction is not a part of the Allgrove
syndrome, TSH levels can be influenced by coexisting adrenal
insufficiency. Congenital hypothyroidism [25] was reported in
association with Allgrove syndrome. Interestingly our case was
also diagnosed to have primary hypothyroidism during her initial
presentation.
ALADIN, a 546 amino acid long protein, is ubiquitously present
in cytoplasm [18]. More than 65 mutations have been reported in
16 exons of the gene. Depending upon the resultant product, the
mutations were divided into truncating (T) and nontruncanting (NT)
protein groups. While patients in the T group show higher prevalence
of AI, the NT group shows a higher prevalence of ND. But the T
group tends to have an early onset of ND, if present [2]. The mutation
in our case is located in exon 6 of AAAS gene, whereas a similar
mutation was previously reported in exon 7 in another database [18].
Allgrove syndrome has so far been reported through case reports. No
genotypic-phenotypic correlation observed [16].
Conclusion
Despite with many case reports and reviews, Allgrove syndrome
continues to be diagnosed late in the majority of patients. A
high index of suspicion is needed for every symptom of alacrima
and Glucocorticoids insufficiency. Thorough biochemical and
radiological investigations are strongly recommended in all suspected
cases. Despite sparse genotypic-phenotypic correlation, patients
should be tested for genetic diagnosis, which can be useful for genetic
counseling as well as follow-up planning. In view of the vast array
of presentations, more research should be carried out for better
understanding of the disease.
References
Citation
Vasishta Reddy S, Kasyapa Jannabhatla VB, Tirupathe S, Bhimeswara Rao P. Allgrove Syndrome without Achalasia: A Rare Case Report and Brief Review of Literature. Indian J Appl Radiol. 2023;9(1): 184.