Case Report
Joubert syndrome: a Rare Radiological Case in Tertiary Care Hospital
Borikar BY, Sable D and Tayade A
Department of Radiodiagnosis, Mahatma Gandhi Institute of Medical Sciences, Sevagram, Maharashtra, India
*Corresponding author: Borikar BY, Department of Radiodiagnosis, Mahatma Gandhi Institute of Medical Sciences,
Sevagram, Maharashtra, India Email: bhagyashri.4ever4u@gmail.com
Copyright: © 2022 Borikar BY, et al. This is an open access article distributed under the Creative Commons Attribution
License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is
properly cited.
Article Information: Submission: 02/03/2022; Accepted: 22/04/2022; Published: 26/04/2022
Abstract
Joubert syndrome is an unprecedented autosomal recessive neuro developmental disorder characterised via way of means of atypical respiratory styles
composed of episodic tachypnea/apnea, hypotonia, ataxia, developmental delay, highbrow impairment, ocular impairment, renal cysts, and hepatic fibrosis.
We record the case of 17 months old boy who presented with fever with cough for 8 days with respiratory distress and rapid noisy breathing, Crepts,
global developmental delay with audio-visual impairment and moderate to severe hearing loss, chronic kidney disease, hypotonia in all four limbs, and plantar
extensors in bilateral lower limbs with pendular nystagmus. Magnetic resonance imaging showing molar teeth signal and a batwing look of the fourth and
absence of the characteristic “focal red dot” deep within the interpeduncular fissure on colour-coded FA-maps.
Keywords
Case Report; Joubert Syndrome and related disorders; clinical and neurological signs; MRI; Diffusion tensor imaging
Introduction
Joubert syndrome (JS) is an autosomal recessive neurological
sickness named after Marie Joubert in 1968 [1]. Joubert syndromeassociated
disorders are assessed into six phenotypes [1,,7]. Such entities
encompass Joubert syndrome with renal defect, Joubert syndrome with
ocular defect (natural JS), Joubert syndrome with oculorenal defects,
Joubert syndrome with hepatic defects, and JS with oro-facio-digital
defects [4]. It offers with unusual oculomotor findings, hypotonia,
ataxia, breathing dysregulation, and developmental retardation as a
result of abnormalities of the cerebellum and brainstem [3-5]. Classic
JS is characterised via the triad of hypotonia, developmental delays,
and pathognomic brainstem and cerebellar malformation known as
the molar teeth sign (MTS) [1,,6]. However, extra recently, the Joubert
syndrome-associated disorders (JSRD) has been followed to explain
formerly wonderful pathological entities with the neuroradiological
characteristic of MTS at the same time as related to different organ
systems. Based on organ involvement, The common age at prognosis is 33 months, and therefore, JS is to be taken into consideration a
syndrome with various phenotypes accordingly making it hard to
diagnose the correct subtype for the duration of the new child period
[5].
Case presentation
A 4-year-old boy was offered to the radiology branch as a referred
case from the branch of paediatrics, in which he turned into mostly
admitted for cough and fever for 8 days, noisy breathing since 4-5
days, wheezing, bilateral nystagmus, and gaze instability. These signs
and symptoms emerged at six months of age and steadily worsened.
Further wondering the patient’s mom discovered negative and not on
time developmental milestones. He completed rollover at the age of 5
months and had no social smile till 1 year of age. He doesn’t respond
to the light but alerts on sound. His mother had premature rupture of
membranes for 48 hrs and he was delivered through a normal vaginal
delivery and weighed 2.5 kg at birth. Postnatally he was started on
antibiotics because of premature rupture of membranes. USG was suggestive of bilateral echogenic kidneys with left inguinal hernia.
Blood creatinine level 1.4, hence was referred to a higher centre for the
pediatric surgeon’s opinion. Review Ultrasonography was suggestive
of bilateral polycystic kidney Disease with left inguinal hernia. Rest
there was no records of cough, asthma, feeding issue, or respiration
problems. On family history, there was no history of consanguineous
marriage and he was the first child by birth. No different previous
participants of their circle of relatives had been affected.
On bodily exam, the kid has a normal facial appearance and was
thin and fragile. His weight was -2 to -3 Standard deviation (SD) and
his height was at -2 Standard deviation (SD) at the pediatric increase
chart for his age. His mid-upper arm circumference was 12.5 cm and
he was classified as protein-energy malnutrition grade I. All relevant
investigations sent. Reports were suggestive of Hemoglobin 9 mg/dl
with iron deficiency anaemia and Vit D deficiency.
He regarded to be conscious but not interested in his
surroundings. However, while instructed, he turned into not able to
attention to his gaze on unique objects. The child was evaluated for
developmental delay and audio and vision examination. An ocular
exam discovered bilateral pendular nystagmus without myopia. With
BERA, brainstem auditory evoked potential (BAEP) recording shows
evidence of Vth wave formation at 60 dB Bilaterally, hence ENT
opinion was taken and the child was found to be having moderate
to severe hearing loss. The cardiovascular exam proved to be within
normal limits. No presence of any type of murmur was recorded.
A pulmonary exam showed expiratory wheeze with Crepts present
on bilateral lower zones. Examination findings of the cranial nerves,
other than the oculomotor nerve, were within normal limits. The
motor examination discovered hypotonia in all four limbs, Deep
tendon reflexes were not ellicitable in bilateral biceps, triceps, knee &
ankle, plantar- Extensors noted in bilateral lower limbs.
A complete collection of Magnetic resonance imaging (MRI) scans
had been performed at our radiology department for evaluation and
to find out the cause of delayed milestones, hypotonia and bilateral
nystagmus. We performed MR imaging using a 1.5 Tesla MR imaging
unit (Simens magneton Avanto 1.5 T) and acquired the imaging
with a standard 8-channel head coil. Before DTI measurement, we
measured conventional sagittal and axial T2-weighted fast spin-echo
and coronal T1-weighted spin-echo and FLAIR imaging sequences
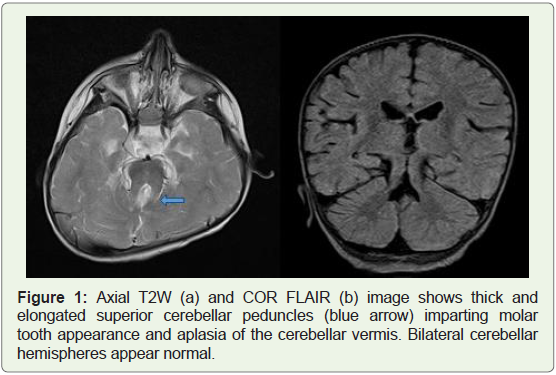
using standard departmental imaging protocols The axial T2-
weighted MRI discovered overall aplasia of the cerebellar vermis with
outstanding, thickened, and elongated advanced cerebellar peduncles
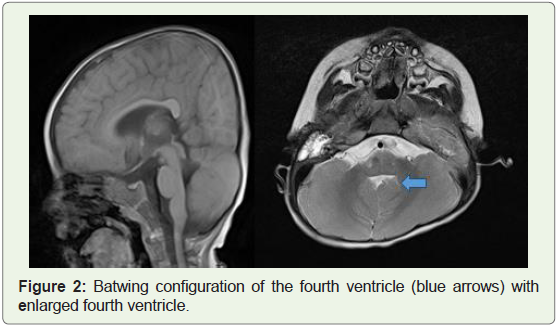
forming a function molar tooth appearance. Furthermore, the fourth
ventricle is regarded as enlarged and triangular, giving it a moderate
batwing appearance (Figures 1 and 2). Based on those clinical findings,
MRI scans, and family history, a prognosis of Joubert syndrome was
made.
For fiber tractography (FT), we transferred the DTI data set
to a personal computer. We performed fiber tractography using
homemade routines based on commercially available image display
software.
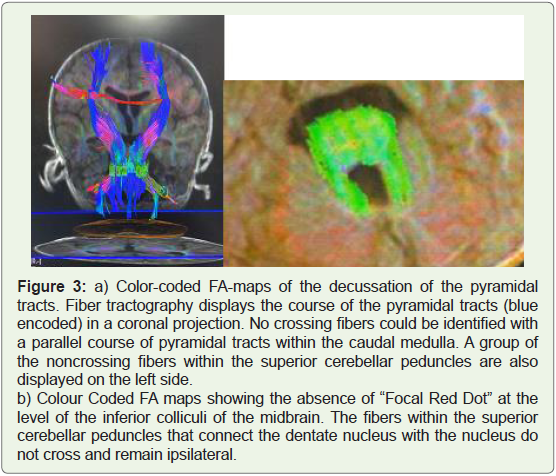
On DTI imaging, the fibers in the superior cerebellar peduncles
were oriented horizontally as represented by a green colour coding
on the FA-maps, instead of slight vertical orientation (blue colour coding) These fibers projected into the red nuclei and thalami
without decussating. The transverse fibers were absent at the level
of the inferior colliculi of the midbrain, with an absence of the
characteristic “Focal red dot” deep within the interpeduncular fissure
on colour-coded Fractional anisotropy (FA) maps. Failure of the
superior cerebellar peduncles to decussate was also demonstrated by
fiber tractography (FT) (Figure 3 and 4).
Discussion
Joubert syndrome is underreported and the incidence ranges
between 1/80000 to 1/100000 live births. JS was originally described
by Marie Joubert in 1968 [1]. Later, Joubert Syndrome-related
disorders (JSRD) were defined based on associated multi-organ
involvement (retinal dystrophy, nephronophthisis, hepatic fibrosis
and polydactyly) [4]. Until 2009, about two hundred instances of
Joubert syndrome were pronounced worldwide, with an additional
12 instances being pronounced to date. Most cases are sporadic, but
some families may show a recessive pattern of inheritance. The correct
diagnosis was often delayed months or years after birth because of its
due to its variable phenotypes, the correct diagnosis of disease was
delayed by months or years even after manifesting the disease at the
neonatal period [3,4].
Joubert syndrome is classified under ciliopathies and to date, 10
causative genes have been discovered. Mutations in the AHI1 and
CEP290 genes are causal in 10-15% and 10% of cases, respectively.
Homozygous deletion of the NPHP1 gene results in 1-2% of cases (2
of original). For normal development and functioning of several cell
types, including retinal photoreceptors, neurons, kidney tubules and
bile ducts, the primary cilia play an important role (Joubert Syndrome
and related disorders) (Table 1 ).
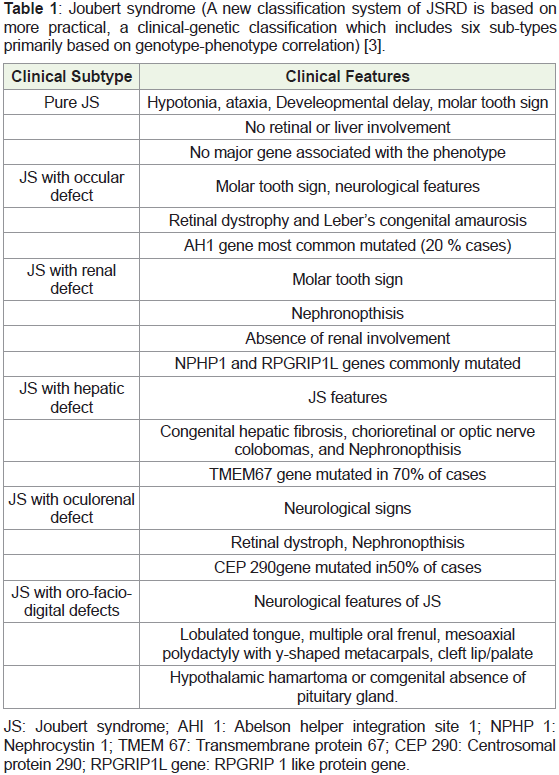
The clinic-pathological manifestations of Joubert syndrome and
Joubert syndrome and related disorders are caused by a defect in
genes encoding for cilium proteins [1,3]. Cilia are either motile or
non-motile organelles that, via ciliogenesis, deliver proteins in both
directions. A defect in ciliogenesis results in the abruption of various
signaling pathways like Wnt, sonic hedgehog, planar cell polarity, and
directional movement.]. Clinically, it is characterized by intellectual
impairment, hypotonia, ataxia, abnormal eye movements, and
abnormal breathing patterns.
Ocular investigations include visible acuity, ocular motility, slit
lamp exam, fundus oculi, and electroretinogram [1]. Standard urine
evaluation with an emphasis on urine unique gravity must be taken
into consideration. An unusual urine unique gravity warrants a
project to take a look at to evaluate the urine concentrating ability.
A belly ultrasound can rule out hepatic fibrosis and proof of renal
structural changes [1]. Though the respiratory dysregulation is more
prominent during the neonatal period diminishing by six months of
age [4,8]. Other findings consist of corpus callosum dysgenesis and
slight lateral ventricular growth [4]. The essential clinical features
of Joubert syndrome are infantile hypotonia, developmental delay,
and one or both of the following: abnormal eye moment and/or
respiratory dysregulation [5,6].
Of the 34 genes regarded to motive Joubert syndrome, 33 are
autosomal recessive, and one is X-linked [6]. The term JS is reserved
for people who meet the diagnostic standards of developmental
delay, extraordinary ocular movements, radiological proof of molar
teeth sign, and cerebellar vermis changes [7]. The terminology JSRD
refers to conditions that have clinical features and radiological signs
of Molar tooth sin along with the involvement of other systems and
organ apart from the central nervous system [7,8].
Radiological findings mainly include characteristic molar tooth signs which help guide the diagnosis of Joubert syndrome [6].
Midline hypoplasia of the cerebellar vermis, incomplete fusion of
the halves of the vermis, abnormally deep interpeduncular fossa, and
thick superior cerebellar peduncles leads to molar tooth sign [4,6-8].
The pontomesencephalic junction is dysplastic with abnormal
decussation of the superior cerebellar peduncle with a marked
decrease in the neurons of the basis pontis and reticular formation
[5]. Histopathological research has shown that the gross appearance
of the brainstem and cerebellum is because of the fragmentation of
the dentate nucleus. Another radiological finding is called a buttock
signal formed due to the absence of the posterior vermian lobe, leaving
the cerebellar hemispheres separated by a cleft. The hypogenesis of
the cerebellar vermis resulting in dilatation of the fourth ventricle
giving batwing or umbrella sign [4,7,8].
Hypotonia and intellectual disability are consistent features
of Joubert syndrome. The altered breathing pattern includes
hyperventilation worsened by stimulation, followed by periods of
apnea or episodic hyperpnea [7]. The physical examination shows
facial dysmorphism including a large head, prominent forehead,
rounded eyebrows, epicanthal folds, ptosis, upturned nose with
evident nostrils, and low-set tilted ear [9]. However, abnormal
respiration is only seen in 68% of cases studied by Pellegrino et
al., and 44% of that Kendall et al. [10,11]. Underlying oculomotor
dysfunction results in abnormal eye movements [9]. Involvement
of the retina leads to fundus flavus, congenital retinal dystrophy,
chorioretinal coloboma, and perimacular and retinal blindness [9].
Other eye findings consist of nystagmus, strabismus, and ptosis.
25% of patients suffer from renal involvement. These patients have
symptoms of polydipsia and polyuria followed by chronic renal
insufficiency manifesting till the second decade of life.
In pregnant females, a prognosis of Joubert syndrome is made
feasible prenatally with the aid of using serial ultrasound imaging
beginning at 11 to 12 weeks gestation. This must be observed with the
aid of using an assessment of cerebellar and fetal anatomy through 20
weeks of gestation and fetal MRI imaging at 20 to 22 weeks gestation
[7].
Lee et al showed thickened superior cerebellar peduncles but did
not demonstrate the absence of decussation of the superior cerebellar
peduncles. Lee et al and Widjaja et al applied diffusion tensor
imaging (DTI), a relatively new MR imaging technique that allows
examination of the course and integrity of white matter tracts in vivo
[12].
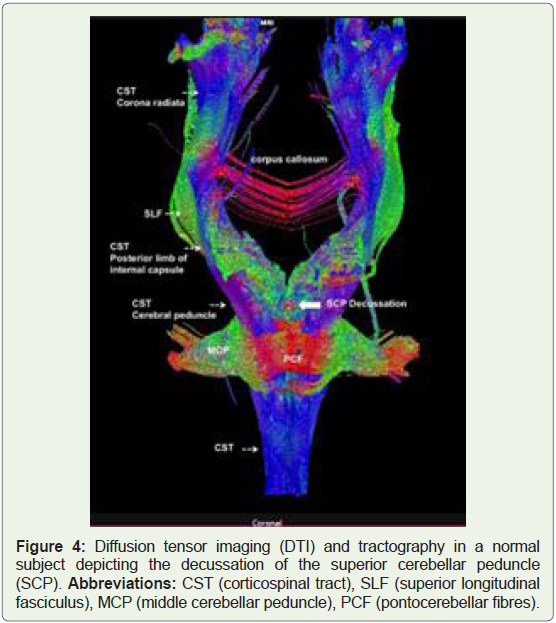
Diffusion-tensor magnetic resonance (MR) imaging (DTI) and
fiber tractography (FT) is new methods that can demonstrate the
orientation and integrity of white matter fibres in vivo [14].
Diffusion tensor imaging (DTI) is an MRI technique that
uses anisotropic diffusion to estimate the axonal (white matter)
organization of the brain. Fibre tractography (FT) is a 3D
reconstruction technique to assess neural tractsusing data collected
by diffusion tensor imaging [14].
MRI are sufficient to confirm or exclude the disease depending
on the detailed clinical history comprising the classical triad of JS and characteristic MTS sign-on After diagnosing with JS/JSRD, the child
must also be evaluated for other organs/system involvement to rule
out multiorgan involvement.
On DTI imaging there shows an almost complete absence of
pyramidal tract decussation in the caudal medulla and abnormal
decussation of the superior cerebellar peduncles with failure of the
superior cerebellar peduncles to decussate in the mesencephalon.
Colour-coded FA-maps are used to evaluate the presence or
absence of a “focal red dot” within the anterior mesencephalon
adjacent to the interpeduncular fossa. The absence of the “focal red
dot” is noted in the absence of decussation of the fibre tracts within
the superior cerebellar peduncles, respectively, as an absence of the
decussation peduncular cerebellarium superior. Similarly, also caudal
medulla oblongata was studied for a “focal red dot” corresponding
to the decussation of the corticospinal tracts. FT was performed to
confirm the findings of colour-coded FA maps and identify a possible
aberrant course of the studied tracts.
Management mainly includes supportive and symptomatic
treatment. Special emphasis should be given to managing respiratory
and feeding. Cognitive problems require a suitable rehabilitation
strategy, and normal follow-up [3]. Due to excessive sensitivity to the
respiratory depressant effects of anaesthetic agents such as opiates
and nitrous oxide these patients require apnea tracking for intensive
care management [4]. The prognosis mainly depends on the type and
extent of organ involvement. Hence, Developmental outcomes may
vary and can result between (a) patients who die young, (b) patients
who survive with developmental delay and visual/motor deficit, (c)
patients whose developmental quotients is in the mildly delayed
range (70 to 80) [9]. Language and motor skills are also delayed in
JS/JSRD patients hence special schooling to learn specific job skills
and to work in a protected environment are also needed [9]. Annual
screening as per diagnostic protocol is advised for such individuals.
Our affected person provided with significant findings of
hypotonia manifested as trouble with neck holding and posture.
However, in our patent, there was no history of parental consanguinity
which is supposed to have a role in the epidemiology of JS as reported
in a study done by İncecik et al. at a rate of 63.6% [8]. Also, he is
presented with bilateral nystagmus and oculomotor dysfunction
with respiratory pattern dysregulation which may or may not worsen
with advancing age. Also, other findings which are consistent with
the diagnosis of Joubert syndrome were the MRI findings of molar
tooth appearance, the batwing appearance of the fourth ventricle, and
hypoplasia of the cerebellar vermis. The patient also presented with
bilateral polycystic kidney disease and bilateral moderate to severe
hearing loss, the diagnosis of JSRD was taken into consideration.
Conclusion
Joubert syndrome clinically presents with respiration
dysregulation, infantile hypotonia, developmental delay, nystagmus,
oculomotor disturbance, and intellectual impairment. The variability
in clinical phenotypes often leads to delayed diagnosis. Diagnosis of
Joubert syndrome requires essential diagnostic criteria in clinical
history along with MRI findings, and a multi-gene panel. The main MRI finding is a molar tooth appearance with concomitant cerebellar
vermis hypoplasia and a batwing configuration of the fourth ventricle.
Management of such cases essentially includes easing respiratory
and feeding difficulties, along with rehabilitation for cognitive and
behavioural difficulties.
Other Differentials of Joubert syndrome could be Joubert
syndrome-related disorders (JSRD), Pontocerebelar dysplasia,
cerebellar hypoplasia syndrome, Congenital Disorders of
Glycosylation.
References
Citation
Borikar BY, Sable D, Tayade A. Joubert syndrome: a Rare Radiological Case in Tertiary Care Hospital. Indian J Appl Radiol. 2022;8(1): 173.