Case Report
A Rare Case of Paediatric Myelin Oligodendrocyte Glycoprotein Antibody- Associated Demyelinating Disease Mimicking Typical Multiple Sclerosis
Das A*, Gautam AK, Agarwal S, Kharkwal R, Issar P, Pal R, Gupta DK
Department of Radio diagnosis, JLN Hospital and Research Centre, Sector 9, Bhilai - 490009, Chattisgarh, India
*Corresponding author: Das A, Department of Radio diagnosis, JLN Hospital and Research Centre, Sector 9, Bhilai -
490009, Chattisgarh , India; Mobile: +91 7489740972; E-mail: mukut016@gmail.com
Copyright: © 2022 Das A, et al. This is an open access article distributed under the Creative Commons Attribution License,
which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Article Information: Submission: 27/12/2021; Accepted: 02/02/2022; Published: 05/02/2022
Abstract
Acquired demyelinating disorders of CNS are rare among the pediatric age group, the common ones among them being ADEM, MOGAD, MS and
NMOSD. Clinical history and MRI of the CNS may reveal some soft indicators that help differentiate the demyelinating disorders, which are extremely
important from the perspective of treatment and prognosis. However, a few overlapping clinical and imaging features may be seen among these disorders.
We present a case of pediatric MOGAD, which had initially mimicked pediatric MS clinically and imaging-wise. We also present his clinico-radiological followup
over a period of 6 years.
Keywords
Pediatric; Demyelinating disorder; Acute disseminated encephalomyelitis; Myelin oligodendrocyte glycoprotein antibody-associated
demyelinating disease; Multiple sclerosis; Neuromyelitis optica spectrum disorder
Introduction
Acquired demyelinating disorders of the central nervous system
(CNS) among the pediatric age group have a rare occurrence, with an
annual incidence of approximately 0.5-1.66 per 100,000 children. The
two most important immunoglobulin G (IgG) antibodies playing a
role in the pathogenesis of these disorders are aquaporin-4 antibody
(AQP-4 Ab) and myelin oligodendrocyte glycoprotein antibody
(MOG- Ab). An MRI of the brain and spinal cord is important in
characterizing the demyelinating lesions, both symptomatic and
subclinical, and in predicting the probability of further recurrences.
Serial MRI may also help in establishing the diagnosis, and
monitoring treatment response [1]. This report describes a rare case
of pediatric myelin oligodendrocyte glycoprotein antibody-associated
demyelinating disease (MOGAD), its clinico-radiological correlation,
and its follow-up over 6 years.
Case Report
March 2015:
An 8-year old male child presented with acute, painless,
diminished vision and was unable to read the question sheet. There
was no history of (h/o) redness in the eye, increased lacrimation, or
watering. No h/o headache, vomiting, seizures, or altered sensorium.
There was no h/o preceding fever, cough, cold, loose stools, or recent
vaccination before the onset of illness. There was no h/o weakness.
There was a past h/o on and off headache since 6 years of age. The
patient was also a known case of (k/c/o) bilateral (B/L) myopia, which
was corrected with refractive lenses. There is a family h/o stroke
in paternal grandmother in 2012. Neurological and fundoscopic
examinations showed normal findings. The child was admitted to
our hospital for 15 days and evaluated. Cerebrospinal fluid (CSF) analysis during hospital stay revealed no cells, protein of 7 mg/dl, and
glucose of 103 mg/dl. Oligoclonal bands (OCB) were detected. CSF
anti-aquaporin 4- immunoglobulin G antibodies (APQ4-IgG) were
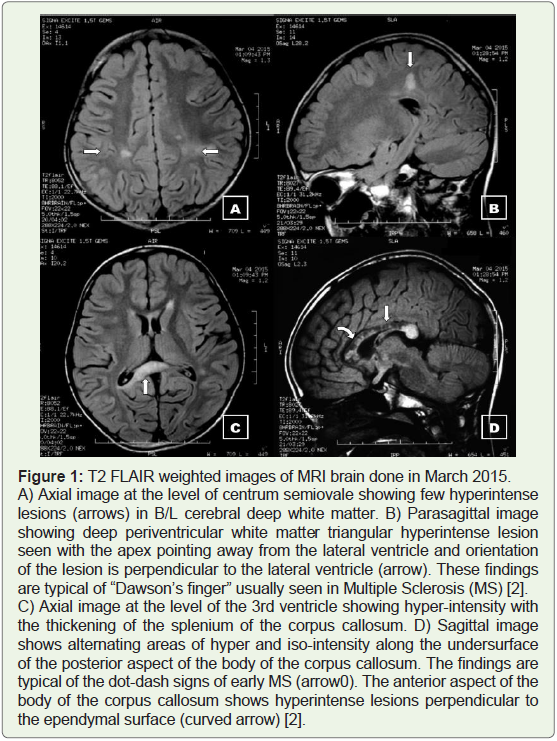
negative. Testing for MOG-Ab was not done.MRI brain (plain + contrast) study revealed multiple
demyelinating T2 FLAIR hyperintense foci in the deep white matter
of B/L cerebral parenchyma including the corpus callosum (Figure 1), typical Dawson fingers, and the ‘dot-dash’ signs of early MS,
thus demonstrating dissemination in space. However, none of them
showed post-contrast enhancement or restriction on DWI. Thus
dissemination in time could not be demonstrated. MRI revealed
no abnormalities in B/L optic nerves. Even though findings that are
typical of Multiple Sclerosis (MS) were seen in this MRI, it did not
fulfil the 2017 revised McDonald criteria [3]. Despite this, in view
of the CSF OCB being positive, a diagnosis of clinically isolated
syndrome/Paediatric MS was made [1].
A diagnosis of ADEM was ruled out as the patient did not have any
recent h/o infection or vaccination. Neuromyelitis Optica Spectrum
Disorder (NMOSD) was unlikely since the patient was APQ4-IgG
negative. The child was treated with pulse methylprednisolone for
5 days followed by oral steroid for 10 days. The child had complete
recovery of vision within 6 days following the onset of illness.
May 2019:
Following the first episode, the child was asymptomatic for four years till May 2019, when he gradually developed weakness in his
upper and lower limbs, with difficulty in walking and in raising his left
arm. There was slurring of speech and change in handwriting over the
next 4 days. There was no h/o headache, blurring of vision, vomiting,
seizures or altered sensorium during this event. On examination, his
vitals were found stable and his higher mental functions were found
normal. On cranial nerve examination, difficulty in blowing mouth
and whistling was found. On sensory examination, there was a loss
of pain sensation in both upper limbs. Power was found to be 4/5 in
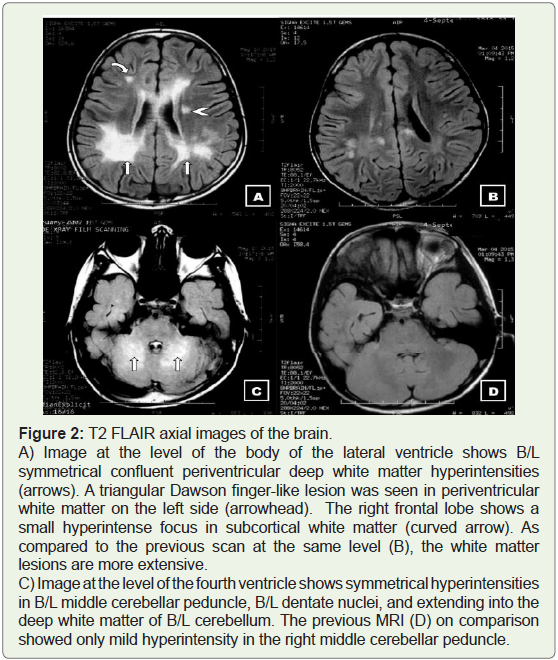
all limbs.MRI Brain (Plain + Contrast) study revealed multiple foci of
T2 FLAIR hyperintensity in the deep white matter of the cerebrum,
confluent hyperintensity in B/L periventricular white matter, B/L
middle cerebellar peduncles, B/L dentate nuclei and extending into
the white matter of B/L cerebellar hemispheres (Figure 2). No focus
of contrast enhancement was seen in the present scan. As compared
to the previous MRI scan done in March 2015, new lesions were
seen. Also, there was more extensive white matter involvement in the
present scan as compared to the previous scan. Thus, dissemination of
demyelinating lesions in both space and time could be substantiated,
which led to the fulfillment of Modified McDonald criteria 2017 [3].
In view of a relapsing attack and a positive OCB in CSF, a diagnosis of
MS was made. However, pediatric MS has a rare incidence. Moreover,
B/L symmetrical cerebellar peduncle involvement along with B/L
confluent periventricular white matter involvement in the pediatric
population is an atypical finding in multiple sclerosis, and more in
favor of MOGAD [4]. Phenotypical similarity in MRI findings may
also be seen in pediatric leucodystrophies [5]. Spinal cord MRI
screening revealed no lesions.
The child was admitted for one week and treated with pulse
methylprednisolone for 5 days followed by oral prednisolone for 8-10
days. There was complete recovery of limb weakness by the third day
following the initiation of treatment.
July 2019:
The child presented with similar complaints and clinical
findings as of those in May 2019. The neuromotor symptoms
were predominantly on the left side of the body. MRI Brain (Plain
+ Contrast) study revealed bilateral white matter T2 FLAIR
hyperintense demyelinating lesions, a few of them showing contrast
enhancements suggestive of acute lesions (Figure 3). These acute
lesions were more concentrated on the right brain, thus correlating
with the left-sided neuromotor symptoms. An open ring pattern of
enhancement was seen in the right middle cerebellar peduncle lesion
with corresponding hyperintensity on T2 FLAIR images. Spinal
cord MRI screening, once again, revealed no demyelinating lesion.
The child was again treated with pulse methylprednisolone and oral
steroids which showed complete recovery within a few days. The
child was evaluated for the above symptoms in September 2019 at a
referral hospital when he was found to be positive for MOG antibody.
A diagnosis of MOGAD was made and was treated with a schedule
of tapering oral steroids for 3 months and an escalating schedule of
mycophenolate.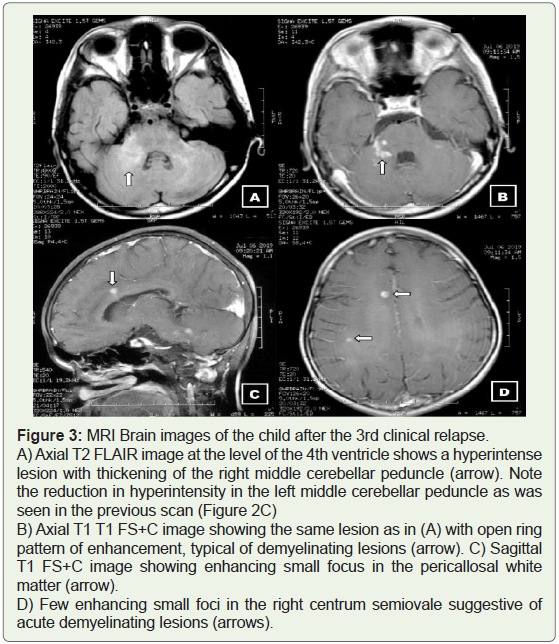
January 2020:
The child again developed gradual onset of weakness in his leftsided
upper and lower limbs, with occasional falls while walking.
There is history of difficulty in grasping objects with the left hand,
and slippage of chappals from the left foot. There is also a history of slurring of speech and change in handwriting, which worsened over
the next 3 days. There is also h/o mild blurring of vision in his left eye
during this event. There is no h/o fever headache, altered sensorium
or seizure. The child was evaluated at the referral hospital for the
above symptoms. On examination, cerebellar signs such as dysmetria
were present (left more than right) and incoordination in the heelshin
test. Pyramidal signs on the left were seen. Visual Evoked
Potential (VEP) test showed left anterior optic pathway dysfunction.
Somatosensory Evoked Potential (SSEP) test done by stimulating the
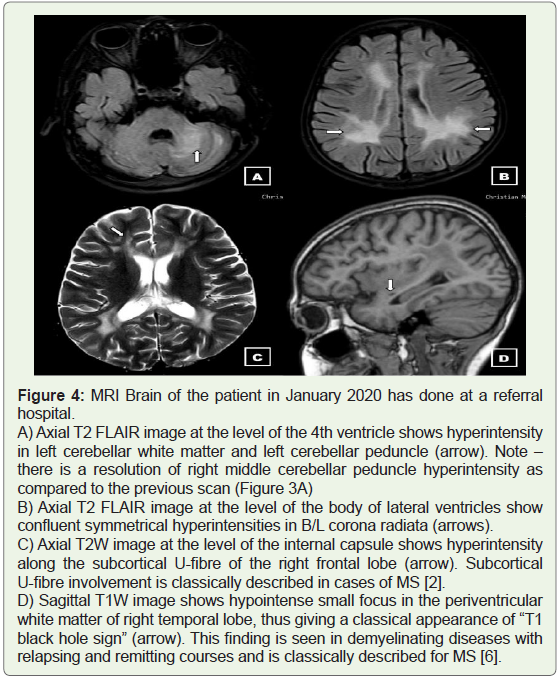
left tibia did not evoke right sensory cortical potentials.MRI Brain (Plain+Contrast) study revealed confluent areas of
T2 hyperintensities in the periventricular white matter and the deep
white matter of frontal and parietal lobes (Figure 4). Left cerebellar
white matter and left middle cerebellar peduncle showed mild
expansion and T2 hyperintensity, which correlated clinically with
cerebellar signs. There was no contrast enhancement or diffusion
restriction. Classical “T1 black hole sign” was seen in right temporal
lobe white matter, which is typical for MS. Since the patient was
having recurrent sensorimotor deficits predominantly on the left side,
3D TOF (Time of Flight) MR Angiography was performed, which
ruled out any vascular abnormality. Spinal cord screening once again
revealed no lesion. The child was given pulse methylprednisolone
for 5 days, followed by rituximab. The patient was discharged on a
schedule of tapering dose of oral prednisolone, followed by prolonged
maintenance on low-dose prednisolone.
February 2021:
A follow-up MRI Brain (Plain + Contrast) was done for the child who had been asymptomatic for the past one year. The findings were
similar to the previous scan, with no significant radiological disease
progression. Few hyper-enhancing foci were seen in the right peritrigonal
white matter suggestive of an acute demyelinating lesion.
The MRI findings are shown in Figure 5. Spinal cord MRI screening
showed no demyelinating lesion.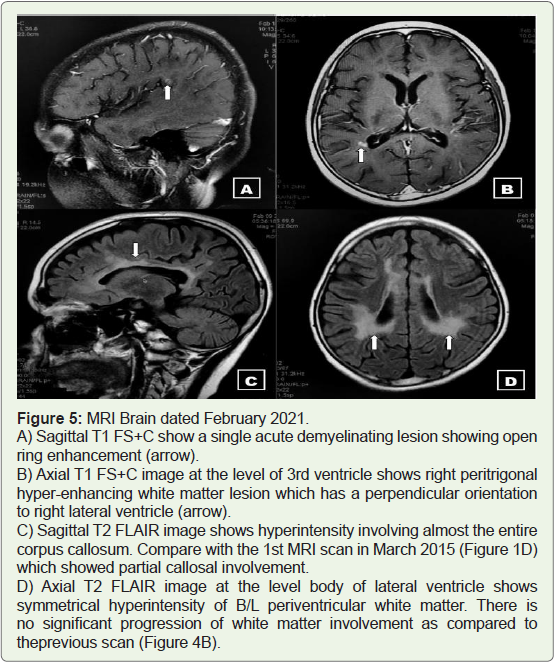
Discussion
Myelin oligodendrocyte glycoprotein (MOG) is a specific protein
found in the outer layers of myelin sheath in the CNS. Earlier, MOG
was associated with MS as per animal model studies [7]. Recent
advances have shown MOG antibodies to be more closely associated
with ADEM and AQP4-Ab negative NMOSD [8,9], rather than MS.
Also, CNS imaging has shown many differences in the phenotype of
MOG antibody-positive patients from AQP4-Ab positive NMOSD
and MS [6,10]. These have lead to a conclusion in recent times that
MOGAD is a separate clinical entity.
In the case of this child, with MRI showing multiple demyelinating
lesions, MOGAD and MS were considered as differentials based on
clinical and radiological findings. ADEM was not considered since
the patient had no h/o recent vaccination or infection. The discussion
on differentials is summarized in Table 1.
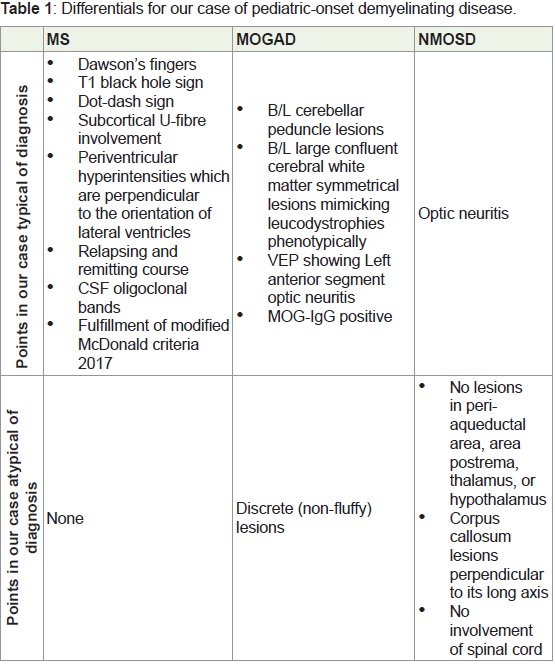
Table 1 show that our case had multiple findings in favor of MS as
well as MOGAD. Our case did not fulfil the international criteria for
MOG-IgG testing [11]. Yet, the positivity of the antibody test clinched
the diagnosis of MOGAD. Ramnathan et al. concluded that there is
a significant overlap of radiological findings in MS and MOGAD
[12]. They recommended testing of all pediatric-onset demyelinating disorders for MOG-IgG due to the high pretest probability of
MOGAD in this population.
Few salient features that were noted in our case and worth
mentioning were as follows:
Between the 1st and 2nd episodes of the disease, there was
an asymptomatic period of 4 years. However, a comparison
of both the MRIs showed significant progression in the
involvement of B/L cerebral white matter. We may conclude
that there was an ongoing process of subclinical demyelination
during these years.
Initial disease morphology was classically that of MS, but later
follow up MRI started showing features of MOGAD
The periventricular white matter had more extensive
involvement in parietal lobes compared to frontal or temporal
lobes.
Most relapses had predominant ipsilateral and unilateral
(left-sided) neuromotor deficits.
Our case mostly did not show a strong correlation between
clinical deficits and imaging findings.
Imaging findings did not show changes in the optic nerve
when clinically optic neuritis was present.
It is extremely important to identify MOGAD apart from MS as
there is a difference in the management of the two disease entities.
Chronic immune suppression may be recommended for patients
with a relapsing disease or who develop steroid dependence, especially if there is incomplete recovery. Commonly used chronic
immunosuppressive agents include mycophenolate mofetil,
azathioprine, rituximab, and monthly IVIG. Most multiple sclerosis
disease-modifying agents are not effective in preventing attacks of
MOGAD [13].
Conclusion
Based on our experience with this case and its continuous followup,
we would like to recommend pediatric-onset demyelinating
disorder for MOG-IgG testing as imaging findings alone can mimic
MS.
References
Citation
Das A, Gautam AK, Agarwal S, Kharkwal R, Issar P, et al. A Rare Case of Paediatric Myelin Oligodendrocyte Glycoprotein Antibody- Associated Demyelinating Disease Mimicking Typical Multiple Sclerosis. Indian J Appl Radiol. 2022;8(1): 171.