Case Report
Anomalous Origin of Left Coronary Artery from Pulmonary Artery Peculiar Presentation in Adulthood: CT Angiographic Features and Comprehensive Review of Literature
Chaturvedy KR1, Raichandani S2*, Ramanand G1 and Latif MA3
1Department of Radiodiagnosis, Dr. S. N. Medical College, India
2Dr. S. N. Medical College, India
3Department of Diagnostic Radiology, Mount Sinai Medical Center, USA
Corresponding author: Raichandani S, Dr. S. N. Medical College, 23, Pal Link Road, Jodhpur, Rajasthan-342008, India, Tel no: +91-9413610800; E-mail: surbhiraichandani@gmail.com
Citation: Chaturvedy KR, Raichandani S, Ramanand G, Latif MA. Anomalous Origin of Left Coronary Artery from Pulmonary Artery Peculiar Presentation in Adulthood: CT Angiographic Features and Comprehensive Review of Literature. Indian J Appl Radiol. 2018;4(1): 124.
Copyright ©2018 Chaturvedy KR, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Indian Journal of Applied Radiology | Volume: 4, Issue: 1
Submission: 17/05/2018; Accepted: 09/07/2018; Published: 12/07/2018
Abstract
Anomalous Origin of the Left Coronary Artery from the Pulmonary Artery (ALCAPA) is a rare congenital coronary anomaly. Coronary anomalies have a broad symptomatology spectrum ranging from asymptomatic with a benign course to severely symptomatic with chest pain to sudden cardiac death. ALCAPA is one such type, associated with early infant mortality and sudden cardiac death in adults. ALCAPA has conventionally been diagnosed by angiography or autopsy; however, the availability of robust, and non-invasive diagnostic imaging modalities like Cardiac Computed Tomography (CCT) and Magnetic Resonance Imaging (MRI) has resulted in easier evaluation of the coronary anatomy by direct visualization of the origin of the left coronary artery from the pulmonary artery. ALCAPA is more accurately diagnosed by two-dimensional and color Doppler echocardiography or Multi-Detector CT. It is important to be cognizant of this unusual but serious cardiac malformation in imaging studies of the heart to prevent avoidable death by instituting timely surgical intervention.
Keywords: Anomalous Left Coronary Artery arising from Pulmonary Artery (ALCAPA); Coronary anomaly; Bland white garland syndrome; Sudden cardiac death
Introduction
Coronary anomalies are uncommon, presenting in 0.3% to 5.6% of the population with most patients reportedly asymptomatic [1]. However, it is important to recognize that they can present with early mortality in infants and young adults from congestive heart failure in the former, to sudden cardiac deaths during strenuous physical activity in the latter. Without surgery, a majority of infants die in the first year [2]. As it predominantly presents in the first year of life, diagnosis in living adults is extremely rare [3].
In this article, we present the clinical and imaging findings of a 28 year old with dyspnea who was diagnosed with ALCAPA.
Case Report
A 28-year-old mother of two was admitted to our hospital with complaints of syncope and shortness of breath during exercise for over 6 months. She did not have a significant past medical or surgical history and her two pregnancies were uneventful. On physical examination, the chest was clear, S1 was normal, but a soft murmur with split S2 was detected on auscultation. A transthoracic echocardiographic examination was then performed, revealing the presence of an abnormal vessel insertion into the Pulmonary Artery (PA), continuous shunt into PA and inter-coronary collateral signals within the ventricular septum. 2D Echocardiography also showed a dilated left ventricle with moderate LV dysfunction, mild mitral insufficiency and decreased ejection fraction. Additionally, a sclerosed anterolateral papillary muscle was noted, raising the suspicion for a diagnosis of ALCAPA.
The patient was then referred to MDCT to definitely diagnose suspected coronary artery anomaly. In the CT angiography, right ventricular outlet revealed narrowing anteroposteriorly with stenosis of the subvalvular/infundibular region. MDCT images also demonstrated LCA arising from the inferolateral aspect of the MPA 1.5 cm above the pulmonary valves with it showing reduced contrast opacification compared to the MPA which was suggestive of retrograde blood flow toward the PA or “pulmonary steal phenomenon”. Also CT angiography exhibited an enormously dilated, tortuous and dominant Right Coronary Artery (RCA) emerging from the right aortic sinus with multiple collateral vessels feeding into the left coronary system along the lateral wall of the left ventricle and the interventricular septal region, which was suggestive of a left to right shunt or “coronary steal phenomenon”. The left Anterior Descending Artery (LAD) and Left Circumflex Artery (LCX) were seen branching from the LCA. Both LAD, LCX and their diagonal and obtuse branches were dilated. The posterior interatrial segments of RCA and PDA were noted to be small in size and most of the ventricular walls were being supplied from the extensive intercoronary collateral branching along both anterior and posterior ventricular walls, as well as the interventricular septum (Figures 1-4).
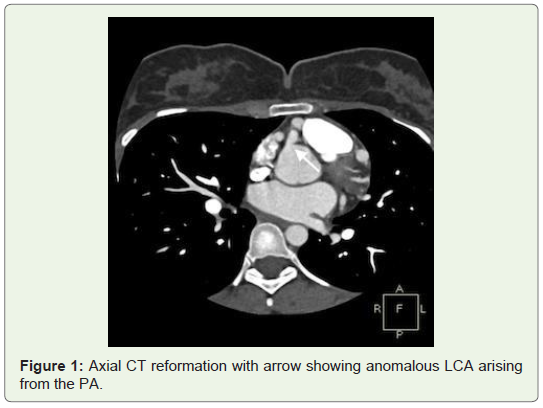
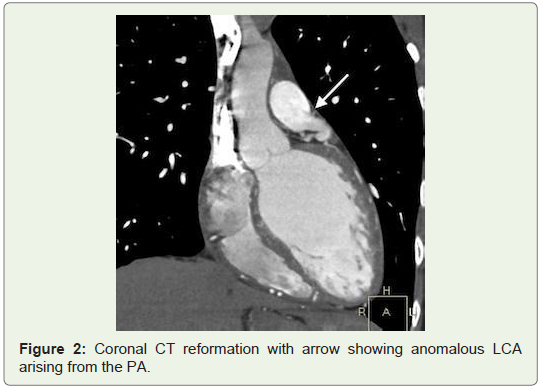
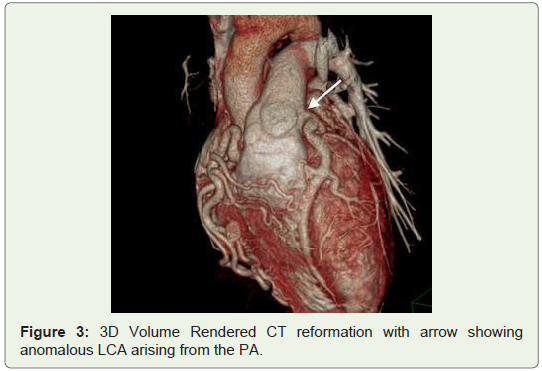
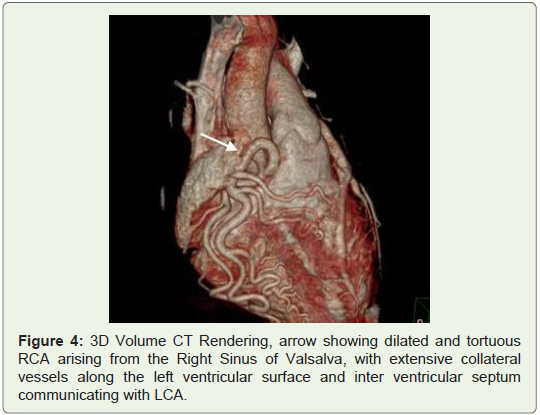
Discussion
ALCAPA is a rare congenital anomaly in which the Left Coronary Artery (LCA) branches off the pulmonary artery instead of the left coronary sinus of the Ascending Aorta.
Comprising 0.25 - 0.5% of all congenital cardiac anomalies [4], it has an estimated prevalence of one in 300,000 live births with a greater than 2:1 predominance of females but no predilection for race [1-3]. ALCAPA is usually an isolated cardiac anomaly and was first described in 1866 by Brooks [2,4].
The earliest clinical description in conjunction with autopsy findings were reported by Bland, et al. hence named ‘Bland-White-Garland syndrome’ [6]. Later on in 1962, ALCAPA was reported in a series of 58 post-mortem studies carried out by Fontana and Edwards and these autopsies demonstrated that most patients had died at a younger age [7].
Abnormal embryonic development may lead to positional abnormalities of the coronary arteries [1]. This congenital cardiac anomaly may result from: abnormal septation of the conotruncus into the aorta and pulmonary artery, or persistence of the pulmonary buds together with involution of the aortic buds that eventually form the coronary arteries. The coronary artery buds appear on about the 12th day of life after the division of the truncus arteriosus, which leads to the separation of the aorta and pulmonary artery [8]. Coronary artery anomalies can arise secondary to either malrotation of the spiral septum dividing the truncus or malpositioning of the coronary buds themselves. Soloff et al has described the four possible anomalous coronary artery connections to the pulmonary artery [9]. The most common being ALCAPA, followed by Anomalous Right Coronary Artery from the Pulmonary Artery (ARCAPA), origin of both coronaries from the pulmonary trunk, and origin of an accessory coronary artery from the pulmonary trunk. The relatively high incidence of ALCAPA is explained by the proximity of the left coronary bud to the pulmonary artery sinus.
Symptoms can appear within the first 2 months of life. ALCAPA does not present prenatally because of favorable fetal physiology accounted for by a pulmonary artery pressure equaling the systemic pressure, allowing for satisfactory myocardial perfusion from the pulmonary artery through the anomalous coronary artery [2]. Physiologic pulmonary hypertension during the first month of life, also tends to preserve antegrade blood flow within the left coronary artery, and infants usually remain asymptomatic [1,4]. Soon afterwards though, pulmonary vascular resistance drops, resulting in low pressure perfusion of the LCA, and this combined with the poor oxygen content in the pulmonary artery leads to a state of relative ischemia in the myocardial areas supplied by the abnormally arising LCA.
The pressure in the RCA, originating correctly from the aortic sinus, soon exceeds that of the anomalous LCA and collateral circulation is stimulated, which ultimately, can lead to blood flowing from the RCA into the LCA (retrograde) and into the pulmonary artery, thus forming a left-to-right shunt, and also known as coronary artery steal [10].
Further increase in myocardial oxygen demand culminates in infarction of the anterolateral left ventricular free wall, often with resultant mitral valve insufficiency secondary to a dilated mitral ring or infarction of the papillary muscle, eventually producing a picture of congestive heart failure [2]. Initially symptoms are exertional - pallor, diaphoresis, tachycardia and/or tachypnea - during feeding or crying. Inadequate myocardial perfusion likely causes significant chest pain, which may be misinterpreted as routine infantile colic [11]. On physical examination cardiac murmurs are a frequent finding [3]. Usually, the disease presents in infancy with 90% infants at risk of death within the 1st year of life without corrective surgery [1,3,4]. Sometimes however, it manifests in adults resulting in chronic myocardial ischaemia and dysrhythmias - a cause of sudden cardiac death in this population demographic [12]. When collaterals are adequate, symptoms can be absent or relatively minor, allowing growth into adulthood. Adults can sometimes be asymptomatic or, more commonly, can have a variety of symptoms such as syncope, chest pain, and sudden cardiac death due to malignant arrhythmias, especially during exercise [2,13].
In a review of 25 infant and adult cases, Kaunitz, et al. confirmed that symptom onset often coincided with closure of the ductus arteriosus 2 months after birth [14]. He also noted in the adult cases, RCA dilation and significant development of collateralization from the RCA to the LCA. This became the first distinction proposed between infants who die in the first year and those who survive to adulthood [13]. Other factors that may lead to survival beyond infancy include an alteration in hemodynamics that encourages antegrade blood flow into the left coronary arterial tree, and a reduction in the area of the left ventricular myocardium supplied by the LCA [4].
The above findings were confirmed in our case as extensive collateral vasculature between the RCA and LCA supplied normally oxygenated blood to the LCA territory and was the mechanism of survival till adulthood. This finding was also presumed to be responsible for the patient’s two fairly uneventful pregnancies.
Early diagnosis is very important because surgical treatment with optimal timing generally results in an excellent prognosis as a result of both rapid detection using echocardiography with color flow mapping and improvements in surgical techniques, including myocardial preservation. Two-dimensional and color Doppler echocardiography is often diagnostic, and should be performed first. Several studies have established diagnostic criteria for ALCAPA using echocardiography. This includes identification of a dilated RCA, retrograde Doppler flow from the LCA to PA, and prominent septal flow from collateralization [3]. Conventional angiography is a traditional technique that has been used for many decades to diagnose coronary artery anomalies [1]. However, MDCT has recently become accepted as the gold standard for evaluation of coronary anomalies, as it allows for accurate and noninvasive depiction of coronary anomalies and has been determined to be superior to conventional angiography for delineating the origin, course and termination of abnormal coronary arteries [15], thus aiding in definitively diagnosing ALCAPA [13]. MDCT is fast and offers excellent spatial resolution, which is required to assess small vessels such as the coronary arteries. The short investigation time, relative noninvasiveness, simple preparation and minimal aftercare make MDCT coronary angiography advantageous over conventional coronary angiography. The main disadvantages of MDCT over angiography are its relatively high radiation dose and its inability to assess flow [13].
ECG-gated cardiac CT allows direct visualization of anomalous left main coronary arterial origin from the posterior aspect of the pulmonary artery. The right coronary artery may be unusually dilated and tortuous with evidence of collateral formation. Intercoronary collateral arteries along the external surface of the heart or within the interventricular septum may also be seen [3,4].
In our patient, CT angiography provided direct visualization of the LCA arising from the main pulmonary artery along with extensive intercoronary collateralization along the external cardiac ventricular surface and the interventricular septum, which was diagnostic of ALCAPA syndrome.
Other modalities that can be used in the diagnosis include a Chest X-ray, which often reveals cardiomegaly, with or without pulmonary venous congestion [2]. ALCAPA is among the differential diagnoses for massive cardiomegaly in the newborn period, a finding that is one of the classic features of this entity at presentation.
As with CT, MR imaging is highly accurate in the detection of the proximal course of anomalous coronary arteries with an additional advantage of demonstrating communication and flow from the LCA into the PA, and is useful in assessing mitral valvular function as well. Delayed gadolinium enhancement may also help determine myocardial viability. MR imaging also does not make use of ionizing radiation, but the main disadvantages of MR imaging in comparison with CT are its relatively long examination times and low spatial resolution. Cardiac CT and MRI also find a use in offering prognostic information and risk stratification besides being utilized for long term follow up imaging [13].
In general, once the diagnosis of ALCAPA is established, early surgical repair is undertaken to prevent potential future complications [4]. Current surgical procedures are directed at establishing revascularization by creating a dual coronary perfusion system [1]. In the infantile type, this is done by either direct reimplantation of the anomalous LCA into the aorta or creating an aortopulmonary window and an intrapulmonary tunnel extending from the anomalous ostium to the window, the Takeuchi procedure [1,2]. In the adult type, a left subclavian arterycoronary artery anastomosis can be established or the LCA can be ligated followed by placement of a saphenous venous graft. Ultimately, by establishing a patent two-coronary artery system, most patients experience normalization of left ventricular systolic function, thereby improving long-term survival. Postoperatively, diuretics, and after load reduction may be necessary until there is significant improvement in left ventricular systolic and diastolic function with resolution of mitral valve insufficiency. Although unusual, there remains a risk of cardiac dysrhythmia secondary to preoperative myocardial ischemia or infarction, requiring steady monitoring in the postoperative period.
Estimated long-term survival at 20 years was recently shown to be 94.8% post surgical repair [13].
In the present case, the patient was offered surgery for which she and her relatives expressed refusal, following which the patient was lost to follow up.
Conclusion
Our case is unique not only because the patient survived to adulthood, but also because she underwent two relatively uneventful pregnancies without significant symptoms. CT angiography revealed an anomalous LCA arising from the MPA, which was the diagnostic hallmark for ALCAPA syndrome.
ALCAPA is a rare but life-threatening condition with an excellent prognosis if early diagnosis and appropriate surgical treatment is instituted. Thus, it should always be considered in the radiological evaluation of an infant with cardiomegaly or an adult with ectatic coronaries encountered in CT or MR imaging.
References
- Lardhi AA (2010) Anomalous origin of left coronary artery from pulmonary artery: A rare cause of myocardial infarction in children. J Family Community Med 17: 113-116.
- Khanna A, Torigian DA, Ferrari VA, Bross RJ, Rosen MA (2005) Anomalous origin of the left coronary artery from the pulmonary artery in adulthood on CT and MRI. AJR Am J Roentgenol 185: 326-329.
- Arciniegas E, Farooki ZQ, Hakimi M, Green EW (1980) Management of anomalous left coronary artery from the pulmonary artery. Circulation 62: 1180-1189.
- Bland EF, White PD, Garland J (1933) Congenital anomalies of the coronary arteries: report of an unusual case associated with cardiac hypertrophy. Am Heart J 8: 787-801.
- Fontana RS, Edwards JE (1962) Congenital cardiac disease: a Review of 357 case studies pathologically. WB Saunders, pp: 291.
- Yau JM, Singh R, Halpern EJ, Fischman D (2011) Anomalous origin of the left coronary artery from the pulmonary artery in adults: A comprehensive review of 151 adult cases and a new diagnosis in a 53-year-old woman. Clin Cardiol 34: 204-210.
- Akgun V, Battal B, Karaman B, Ors F, Saglam M, et al. (2010) A case of anomalous left coronary artery arising from the pulmonary artery in adulthood: multidetector computed tomography coronary angiography findings. Eurasian J Med 42: 100-102.
- Danias PG, Stuber M, McConnell MV, Manning WJ (2001) The diagnosis of congenital coronary anomalies with magnetic resonance imaging. Coron Artery Dis 12: 621-626.
- Soloff LA (1942) Anomalous coronary arteries arising from the pulmonary artery. Am Heart J 24: 118-127.
- Edwards JE (1964) The direction of blood flow in coronary arteries arising from the pulmonary trunk. Circulation 29: 163-166.
- Su LS, Burkhart HM, O’Leary PW, Dearani JA (2011) Mitral valve arcade with concomitant anomalous left coronary artery from the pulmonary artery. Ann Thorac Surg 92: e121-e123.
- Peña E, Nguyen ET, Merchant N, Dennie C (2009) ALCAPA syndrome: not just a pediatric disease. Radiographics 29: 553-565.
- Oncel G, Oncel D (2013) Anomalous origin of the left coronary artery from the pulmonary artery: diagnosis with CT Angiography. J Clin Imaging Sci 3: 4.
- Kaunitz PE (1947) Origin of left coronary artery from pulmonary artery: Review of the literature and report of two cases. Am Heart J 33: 182-206.
- Schmitt R, Froehner S, Brunn J, Wagner M, Brunner H, et al. (2005) Congenital anomalies of the coronary arteries: imaging with contrast-enhanced, multidetector computed tomography. Eur Radiol 15: 1110-1121.