Case Report
Case Report: Radiogenomic Insights into Corpus Callosum Dysgenesis with Hypoplastic Septum Pellucidum
Baby NM*
Department of Radiology, Government Medical College, Ernakulam, Kerala, India
*Corresponding author:Nikita Mary Baby, Senior Resident, Department of Radiology Government Medical College, Ernakulam, Kerala, India. E-mail Id: nikita92.nmb@gmail.com
Copyright: ©2025 Baby NM. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Article Information:Submission: 19/09/2024; Accepted: 20/12/2024; Published: 02/01/2025
Abstract
This case report presents a radiogenomic approach to a patient with MRI findings of Corpus Callosum Dysgenesis (CCD) and Hypoplastic Septum Pellucidum (HSP). By meticulously observing and documenting radiological data, we integrate these findings to identify the causative genetic mutations, predict the neurodevelopmental course, and guide treatment planning. This case and approach highlight the potential to leverage radiogenomics using routine MRI findings to improve therapeutic strategies.
Keywords:Corpus Callosum Dysgenesis; Hypoplastic Septum Pellucidum; Radiogenomics; Genetic Mutations; Neurodevelopmental Disorders; Magnetic Resonance Imaging (MRI).
Introduction
Corpus Callosum Dysgenesis (CCD) with Hypoplastic Septum
Pellucidum (HSP) is a complex neurodevelopmental condition
with diverse clinical manifestations. Radiogenomics, by integrating
imaging findings with genetic analysis, enhances the understanding
of genotype-phenotype correlations. Careful observation and
documentation of radiological features serve as the cornerstone for
identifying underlying genetic mutations. This report focuses on the
radiogenomic evaluation of CCD and HSP, emphasizing the role
of MRI in guiding genetic testing and personalized care. As noted
by Rudas et al., “Corpus callosum malformations are frequently
associated with other brain anomalies, including ventricular
enlargement and cortical dysgenesis, which underscores the
importance of early genetic evaluation” (Rudas et al., 2024)[1].
Case Presentation
Patient Information
• Age: 7 years
• Gender: Female
A 7-year-old female presented with significant developmental delays affecting cognitive, motor, and language functions.
• Age: 7 years
• Gender: Female
A 7-year-old female presented with significant developmental delays affecting cognitive, motor, and language functions.
Developmental History:
The patient displayed an abnormal developmental trajectory from
infancy, with delayed milestones such as late walking and persistent
poor coordination. Early intervention therapies were initiated but
showed limited improvement. Social challenges were also noted,
including reduced engagement and difficulty communicating with
peers.Family History:
The family history was largely unremarkable for neurological
conditions, except for the patient’s maternal grandmother, who had
developmental delays. This raised the possibility of a hereditary or
carrier genetic condition.The primary concerns included:
• Cognitive impairments: Difficulty with attention, memory, and language acquisition.
• Motor delays: Poor coordination, balance issues, and impaired fine motor skills.
• Speech delays: Challenges in articulation and comprehension.
Neurological Examination:
Key findings from the neurological examination included:• Hypotonia: Decreased muscle tone.
• Motor incoordination: Balance difficulties and fine motor impairments. No seizures or other overt neurological deficits were observed.
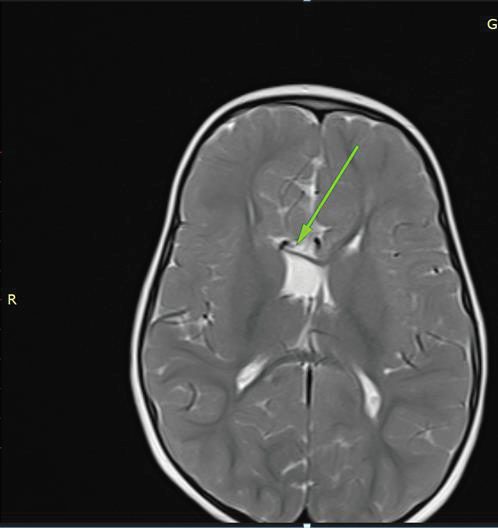
Imaging Findings:
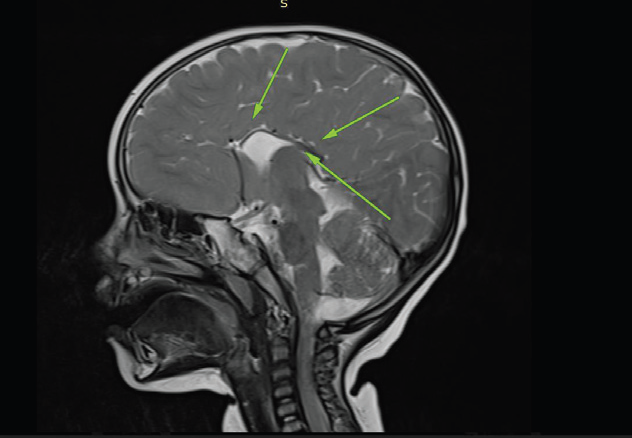
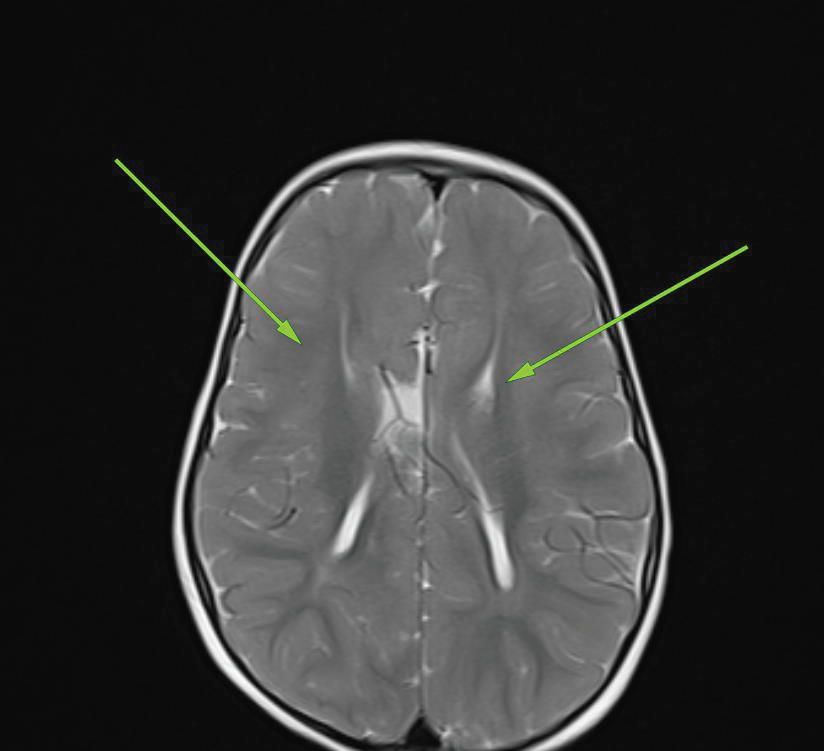
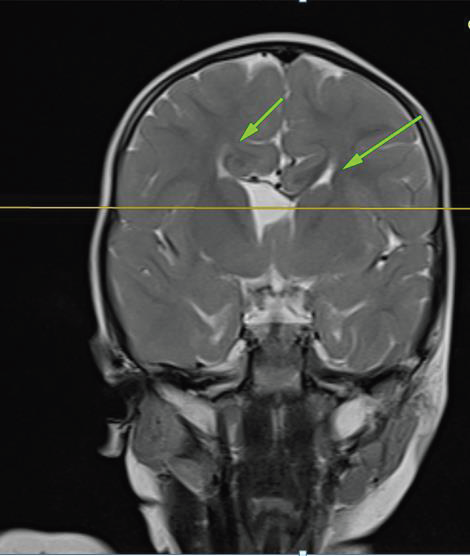
MRI BrainStructural MRI revealed:
• Agenesis of the corpus callosum.
• Hypoplasia of the septum pellucidum.
• Mild ventricular enlargement.
• Pachygyria
CT Scan:
CT confirmed the absence of the corpus callosum and provided
additional details on ventricular morphology.CT scan was performed
to rule out any associated corpus callosal lipoma.Radiogenomic approach:
The imaging findings indicate a disrupted neuronal migration
and midline development.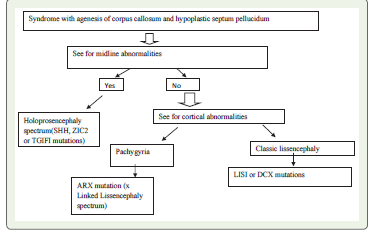
Genetic Analysis:
Genetic TestingWhole-exome sequencing revealed a pathogenic missense mutation in the ARX gene (c.1234T>G), a mutation known to be associated with X-linked lissencephaly and CCD. This genetic finding correlated directly with the radiological abnormalities observed.
Discussion
Radiogenomic Correlation
The ARX mutation identified in this case aligns with hallmark radiological features, including corpus callosum agenesis, hypoplastic septum pellucidum, and pachygyria. As Al-Gazali et al. note, “ARX mutations are involved in a wide range of brain malformations, including lissencephaly and agenesis of the corpus callosum” (Al- Gazali et al., 2008)[2].
The ARX mutation identified in this case aligns with hallmark radiological features, including corpus callosum agenesis, hypoplastic septum pellucidum, and pachygyria. As Al-Gazali et al. note, “ARX mutations are involved in a wide range of brain malformations, including lissencephaly and agenesis of the corpus callosum” (Al- Gazali et al., 2008)[2].
Value of Detailed MRI Observation:
Careful analysis and documentation of MRI findings, such
as recognizing CCD, hypoplasia of the septum pellucidum, and
cortical malformations, prompted targeted genetic testing.
Radiological features serve as a reliable guide for narrowing down
potential genetic etiologies and streamlining the diagnostic process.Impact on Treatment:
Integrating radiogenomic insights enhances management by
tailoring interventions:• Therapies for Cognitive and Motor Development: Early intervention programs focusing on motor coordination and cognitive deficits.
• Seizure Surveillance: Anticipatory monitoring given the association of ARX mutations with epilepsy.
• Genetic Counseling: Recommendations for family planning and testing for carrier status.<
Prioritizing Risk-Specific Interventions:
Radiogenomic data can predict potential comorbidities, allowing
for early intervention:• ARX-related CCD: Focus on motor milestones due to hypotonia and cognitive rehabilitation for attention and memory deficits.
• CCD with lissencephaly: Intensive focus on seizure control, respiratory support, and multidisciplinary therapies for severe developmental delays.
• CCD with pachygyria: Tailored developmental programs addressing moderate delays with a better outlook for independence.
Conclusion
Radiogenomic evaluation begins with meticulous MRI analysis,
serving as a bridge to genetic insights. Only when a radiologist
reports a case with radiogenomics in mind can they help the
geneticist successfully derive the causative mutation. Adopting a
radiogenomic approach to a case is crucial to avoid missing subtle
associated radiological findings that may otherwise be overlooked in
the presence of more obvious and gross abnormalities (satisfaction
of search error). Similarly, reporting negative findings helps exclude
alternative genetic causes.
In this case, observing and documenting subtle imaging
features like pachygyria and mild ventricular enlargement led to the
identification of an ARX mutation. Likewise, documenting the absence
of midline abnormalities associated with the holoprosencephaly
spectrum helps exclude other genetic causes, refining the diagnosis
and guiding personalized care. An astutely reported MRI brain
is a crucial tool for a genetic laboratory to narrow down genetic
mutations in a child with developmental delay. This approach
underscores the potential of radiologists to enhance the understanding
and management of complex neurodevelopmental disorders.
Acknowledgements
I would like to express my gratitude to the Department of
Radiology and the Department of Paediatrics at Government Medical
College, Ernakulam, for their support in the imaging studies and
clinical evaluation of this case. I also thank the patient and her family
for their cooperation and willingness to share clinical information,
which was vital for this report. Special appreciation goes to the
radiology technologists for their assistance with the imaging process,
which played a crucial role in accurate diagnosis and analysis.
Conflicts of Interest:
The author declares no conflicts of interest related to this case report.References
Citation
Baby NM. Case Report: Radiogenomic Insights into Corpus Callosum Dysgenesis with Hypoplastic Septum Pellucidum. Indian J Appl Radiol. 2025;11(1): 203.